by Constantine G. Lyketsos, MD, MHS; Steven D. Targum, MD; Jo Cara Pendergrass, PhD; and Andres M. Lozano, MD, PhD, FRCSC, FRSC
Constantine G. Lyketsos, MD, MHS, is Elizabeth Plank Althouse Professor, Department of Psychiatry and Behavioral Sciences, Johns Hopkins University and Chair of Psychiatry at Johns Hopkins Bayview Medical Center; Steven D. Targum, MD, is Scientific Director at Clintara LLC, Chief Medical Officer at Methylation Sciences Inc., BrainCells Inc., and Functional Neuromodulation Inc., Chief Medical Advisor at Prana Biotechnology Ltd., and a consultant in psychiatry at the Massachusetts General Hospital; Jo Cara Pendergrass, PhD is Vice President of Clinical Operations at Clintara LLC; and Andres M. Lozano MD, PhD, FRCSC, FRSC, is Dan Family Professor and Chairman of Neurosurgery at the University of Toronto and RR Tasker Chair in Functional Neurosurgery (Toronto Western Hospital), and Canada Research Chair in Neuroscience.
Innov Clin Neurosci. 2012;9(11–12):10–17
Funding: There was no funding for the development and writing of this article.
Financial disclosures: Dr. Lyketsos has received grant support (research or CME) from NIMH, NIA, Associated Jewish Federation of Baltimore, Weinberg Foundation, Forest, Glaxo-Smith-Kline, Eisai, Pfizer, Astra-Zeneca, Lilly, Ortho-McNeil, Bristol-Myers, Novartis, National Football League, Elan, Functional Neuromodulation Inc. Dr. Lyketsos has been a consultant/advisor to Astra-Zeneca, Glaxo-Smith Kline, Eisai, Novartis, Forest, Supernus, Adlyfe, Takeda, Wyeth, Lundbeck, Merz, Lilly, Pfizer, Genentech, Elan, NFL Players Association, NFL Benefits Office, Avanir, Zinfandel, and BMS. Dr. Lyketsos has received honoraria or travel support from Pfizer, Forest, Glaxo-Smith Kline, Health Monitor; Dr. Targum has received grant support or has been a consultant or medical advisor to Acumen, Acadia Pharmaceuticals, Alkermes Inc., AstraZeneca, BioMarin, BrainCells Inc., CeNeRx, Cephalon, CTNI MGH, EnVivo Pharmaceuticals, Euthymics, Forest Research, Functional Neuromodulation inc, Eli Lilly and Company, Johnson & Johnson PRD, INC Research, Medtronics, Methylation Sciences Inc., NeoSync, Neurophage, Novartis Pharmaceuticals, Novartis Bioventures, Nupathe, Parexel International, PRA International, Prana Biotechnology Ltd., ReViva, Roche Labs, Sophiris, Sunovion, Takeda, Targacept, Theravance, and Transcept; Dr. Pendergrass has no conflicts relevant to the content of this article; and Dr. Lozano is a consultant to Medtronic, St Jude, and Boston Scientific, co-founder Functional Neuromodulation Inc, and has intellectual property in the field of DBS.
Key Words: Alzheimer’s disease, deep brain stimulation, hippocampus; fornix, neuronal circuitry
Abstract: Recent studies have identified an association between memory deficits and defects of the integrated neuronal cortical areas known collectively as the default mode network. It is conceivable that the amyloid deposition or other molecular abnormalities seen in patients with Alzheimer’s disease may interfere with this network and disrupt neuronal circuits beyond the localized brain areas. Therefore, Alzheimer’s disease may be both a degenerative disease and a broader system-level disorder affecting integrated neuronal pathways involved in memory. In this paper, we describe the rationale and provide some evidence to support the study of deep brain stimulation of the hippocampal fornix as a novel treatment to improve neuronal circuitry within these integrated networks and thereby sustain memory function in early Alzheimer’s disease.
Introduction
Alzheimer’s disease (AD) continues to be a growing public health concern. It is estimated that there will be over 115 million new worldwide cases of AD within the next 40 years, resulting in an overwhelming health and economic burden on society.[1] Given the major public health priority of AD, the Alzheimer’s Association released the National Alzheimer’s Plan, From Act to Action, in May 2012 outlining a national strategy to address AD research, care, and services with the specific goal of finding effective ways to prevent and treat the disease by 2025. AD is characterized by progressive cognitive dysfunction often beginning with an early disturbance of episodic memory and ultimately leading to absolute functional impairment. The apparent pathological processes causing AD include misprocessing of fibrillar amyloid leading to oligomerization, the deposition of amyloid plaque causing a disruption of neural network activity, a loss of synaptic function, and eventual neuronal death.[2,3]
The United States Food and Drug Administration (FDA) has approved three cholinesterase inhibitors and the N-Methyl-D-aspartic acid (NMDA) antagonist memantine for the treatment of AD. In practice, these therapies have had only modest, short-term benefits for clinical symptoms but they continue to be widely used because of a lack of alternative treatments.[4–6] Given the current understanding of the pathological route leading to AD, recent clinical research efforts have focused on the development of several “anti-amyloid” therapies. Attempts have been made to modify the effects of amyloid production, aggregation, clearance, or plaque deposition on neuronal tissue. Unfortunately, most of the recent Phase 2 and 3 trials using immunotherapies, “anti-aggregants,” or related agents have been unsuccessful.[7–10] For instance, postmortem studies in AD patients treated with amyloid immunization found that cognitive dysfunction and brain disease progressed despite evidence that the much of the amyloid had actually cleared from the brain.[11] In sum, these recent clinical trials have had disappointing results and generated more interest in seeking novel approaches to treat this devastating illness.
One novel strategy to treat AD is circuitry-based treatment via deep brain stimulation (DBS). DBS treatment has been used in numerous central nervous system (CNS) disorders, including Parkinson’s disease, epilepsy, treatment-resistant depression, bipolar disorder, anorexia nervosa, obsessive compulsive disorder, Tourette’s syndrome, addiction, pain, obesity, and most recently AD.[12] The National Institutes of Health clinical trial registry (www.clinicaltrials.gov) lists over 150 clinical trials of DBS for conditions across the CNS spectrum.
Clearly, alternative treatment options are needed for AD. This article will describe the rationale for assessing DBS as an innovative strategy for the treatment of early AD.
The role of memory networks in Alzheimer’s disease
The evidence of functional alterations in memory networks is commonly seen in AD patients as well as normal elderly persons who have no cognitive impairment.[13–15] Recent data have shown that both normal elderly individuals and patients with AD have defects in heteromodal interconnected cortical areas known collectively as the “default mode network.”[15,16] Young adults show correlated activity within this default network during resting states and deactivation of the network when performing many cognitive tasks.16 On the other hand, older adults lose the expected deactivation and toggling of the default network during cognitive tasks.[15,17] These deficits are more pronounced in patients with AD reflecting impairment in their neuronal circuitry. It is conceivable that defects in the default mode network function, as a consequence of amyloid deposition or other mechanisms, may be responsible for some of the multimodal cognitive and behavioral deficits seen in patients with AD.[15,18,19] Further, it is also possible that dysfunction caused by molecular or structural abnormalities in one diseased brain area may secondarily interfere with the activity in other unaffected brain areas whose function is linked within the integrated neuronal circuitry network. Thus, AD may be both a degenerative disease and a system-level disorder affecting several integrated pathways linking select cortical and subcortical areas that typically work in concert to serve aspects of memory and cognition. If this dual etiologic hypothesis is correct, the modulation of neuronal activity within these interconnected dysfunctional networks may sustain or even improve cognitive function in patients with early AD. Structural abnormalities, the fornix, and memory disturbances Neuroimaging studies of AD have demonstrated the presence of structural abnormalities in AD patients that are directly associated with the cognitive and functional disturbances experienced in AD. Neuroimaging research has shown that the neuropathology occurs in widespread brain regions but with an early preference for neural circuitry that affects memory. Regional reduction in glucose utilization in the temporal lobe and posterior cingulate is a common finding in positron emission and single photon emission computerized tomography (PET/SPECT) in early AD, as well as in healthy individuals at genetic risk for AD.[20,22] These brain regions have a propensity for fibrillar amyloid deposition as visualized at autopsy and in vivo using radiotracers, such as the Pittsburgh compound B ([11C]-PiB) in AD patients and non-demented older adults.[23–27] Structural neuroimaging studies have revealed volumetric changes in the entorhinal cortex and hippocampus that predate the development of cognitive symptoms and subsequently track with the progression of disease.[28,29] In fact, recent studies suggest that loss of the integrity of the fornix, a major in- and outflow track to the hippocampus, may be associated with early memory dysfunction. The fornix is a large axonal bundle estimated to have 1.2 million axons in the human.[30] The importance of the fornix in memory function is supported by the observation that fornix lesions in experimental animals and humans produce memory deficits.[30–33] The fornix integrity can be measured by fractional anisotropy (FA) using diffusion tensor imaging (DTI). One study found lower fornix FA predicted later cognitive decline in individuals with amnestic mild cognitive impairment (aMCI) as well as later hippocampal volume loss.[34,35] This finding of an early structural abnormality in the lower fornix that is associated with the eventual development of AD may become a meaningful clinical predictor of later cognitive deterioration. These recent findings clearly link the fornix to the process of cognitive deterioration in AD and to later hippocampal degeneration as well.
Thus, there is accumulating evidence that defects in the function of the default mode network may occur in AD and that the fornix is linked to this process. These observations support the development of circuitry-based treatments for AD using the fornix as a preferential locale for treatment intervention.
Deep Brain Stimulation
DBS is a circuitry-based treatment. DBS was first tested in animal experiments over 70 years ago and was first approved in Europe in 1995 and in the United States in 1997 and 2002 for essential tremor and Parkinson’s Disease, respectively. The therapy currently is licensed in various regions of the world for a range of additional uses, including dystonia, refractory epilepsy, and obsessive compulsive disorder.[36–38] DBS also has been studied in the treatment of treatment-resistant depression, bipolar disorder, anorexia nervosa, obsessive compulsive disorder, Tourette’s syndrome, addiction, pain, and obesity. Interventions in these dysfunctional circuits can have local, trans-synaptic and remote effects,[39,40] and, in some cases, produce striking clinical improvements beyond what is achievable with medications. DBS is now available in most major medical centers. It is estimated that over 85,000 patients have had implants placed and received brain stimulation to date,[41] and that 8,000 to 10,000 new patients are treated each year.
DBS involves the neurosurgical implanting of indwelling electrodes within specific brain circuits to modulate the activity of those circuits. DBS can suppress pathological neuronal activity or drive underactive output. In this way, DBS can modulate brain circuitry activity up or down based upon the specific needs. The target location for electrode placement is most often done by using structural brain imaging, usually computed tomography or magnetic resonance imaging. The electrodes are commonly placed while the patient is fully awake. Microelectrode recordings are used to differentiate structures or nuclei and to refine lead placement as different structures have specific firing patterns or frequencies, thus enabling more precise lead placements for DBS. In an awake subject, the final placement of the electrodes is abetted because the surgeon can gauge the patient’s response to test stimulation. The electrodes are then connected to an implanted pulse generator that is similar to a conventional cardiac pacemaker. The pulse generator is subsequently programmed to deliver continuous stimulation that can last for several years. Both rechargeable and nonrechargeable devices are available. Nonrechargeable devices typically last 4 to 5 years. Once the batteries are depleted, the pulse generator can be replaced in an outpatient procedure without affecting the electrodes. The level of stimulation is adjusted to optimize the desired outcome and minimize unwanted side effects. The activity of an entire interconnected brain network can be manipulated in this way because the current acts locally, at the site of application, and remotely, at sites that are anatomically connected. The risks for DBS neurosurgery are real but relatively low. There are potential physical adverse effects resulting from the neurosurgery and possible psychiatric effects (e.g., hypomania) that require careful monitoring. There is approximately a one-percent incidence of serious intraoperative events (e.g., cerebral hemorrhage or stroke). There is a 5- to 10-percent risk of ongoing complications related to the hardware, such as breakage, malfunction, and infection.[42] There can also be stimulation-related adverse effects (e.g., paraesthesias, dysarthria, and motor contractions) depending on the placement of the electrode. For instance, motor effects or paresthesia would not be expected from surgery in the area of the fornix. These potential adverse effects are usually reversible if the stimulation is reduced or stopped.
DBS: A possible circuitry-based treatment for Alzheimer’s disease
DBS may be a useful circuitry-based treatment for AD if the electrodes are placed in areas of the brain that are implicated in memory, such as the hippocampal fornix (Figure 1). Although DBS of the fornix (DBS-F) is a very novel strategy, it is potentially an important therapeutic approach to slowing AD as it may slow fornix degeneration, influence hippocampal neurogenesis, and sustain cortical neurons and circuitry by releasing trophic factors.
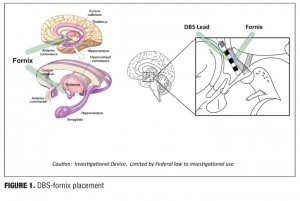
The use of DBS-F to treat AD is supported by rodent studies in which DBS-F treatment revealed improved memory on the Morris water maze task and increased hippocampal neurogenesis based upon histopathological studies.
The animal evidence for DBS-induced neurogenesis is provocative. One study of rodent models found that electrical stimulation of the Papez circuit (of which the fornix and hippocampus are part) using parameters analogous to clinical high frequency DBS reversed memory impairment by corticosterone treatment and produced hippocampal neurogenesis.[43,44] Entorhinal cortex (EC) stimulation increased new granule cell formation in adult mice that were integrated into hippocampal networks engaged by spatial memory in a maturation dependent manner.[45] EC stimulation facilitated spatial memory formation at six weeks but not at one week before or after training. The time delay to special memory is consistent with the time course of incorporation of adult-generated neurons into spatial memory circuits. Importantly, the facilitation of spatial memory by EC stimulation was prevented by blocking neurogenesis. Taken together, these data suggest that DBS can induce neurogenesis, which in turn can facilitate improvement in the integrated neuronal network that affects memory.
A preliminary, open-label study of DBS in early Alzheimer’s disease
The DBS-F treatment strategy for early AD is supported by some promising but preliminary results derived from a small, open-label Phase 1 trial that was conducted in Toronto by Laxton et al.[46] Six AD patients received DBS-F neurosurgical implants and were treated for 12 months with deep brain stimulation. As shown in Figure 2, follow-up PET scan studies over the next year revealed increases in cortical glucose metabolism that were correlated with improved cognitive measures in some of these patients.[46] The finding of increased glucose metabolism is a striking contrast to the well-documented, longitudinal metabolic decline generally seen in AD patients.[20,47] After 12 months, the DBS-F treatment produced a sustained increase in glucose utilization in the posterior cingulate cortex and the precuneus; these are important components of the brain default mode network that are most affected in early AD and where significant amounts of beta-amyloid deposition is known to occur.[33,48,49] Furthermore, using electromagnetic tomography (sLORETA) these investigators demonstrated that DBS drove neural activity in the “memory circuit,” including the entorhinal and hippocampal areas, and activated the default mode network. The activated regions included the anterior cortical (left anterior cingulate, bilateral superior and middle frontal gyrus, and left insula), temporal cortical (bilateral superior, middle and medial temporal cortices), and parietal cortical regions (left precuneus, bilateral posterior cingulate, left fusiform gyrus, right supramarginal gyrus, right inferior parietal lobule) and the bilateral cerebellum.
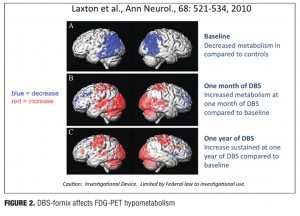
Clinical evaluation using the conventional Alzheimer Disease Assessment Scale cognitive subscale (ADAS-COG) and the Mini Mental State Examination (MMSE) suggested possible slowing in the rate of progressive cognitive decline in these mild AD patients.[50,51] It is important to note that this was a small open-label study without any placebo control group, and the results must be interpreted with caution. Further, these patients had very mild AD at the time of DBS implantation. However, these findings do suggest that the underlying DBS mechanism of action may involve up-regulation of processing, capacity, and/or sustained integrity of these neuronal circuits. Hence, a circuitry-based treatment like DBS may be helpful for patients with early AD.
Current research for DBS in early Alzheimer’s disease Currently, Clinicaltrials.gov lists six distinct clinical trials evaluating the role of DBS in memory disorders being conducted in Europe and the United States. Double-blind studies of patients with DBS implants are possible because the brain stimulation can be turned on or off. Therefore, patients who have had surgical implants can be randomly assigned to “active-on” or “sham” stimulation groups during the double-blind treatment period. One currently enrolling double-blind, “active-on” versus “sham” DBS study (clinicaltrials.gov NCT01608061) is a Phase 2b study for patients with mild AD (defined as meeting diagnostic criteria of probable AD based on the National Institute of Aging Alzheimer’s Association Guidelines, obtaining an ADAS-Cog 11 item score of 12–24, and a CDR Global Score of 0.5–1). All patients will be on standard medical treatment for AD and will be randomly assigned to DBS-F activation either following surgery or after a 12-month delay, and followed for 24 months post-operatively. Comprehensive safety, clinical, cognitive, and neuroimaging assessments will be performed, including serial PET scans of glucose metabolism and magnetic resonance scans of hippocampal volumes/fornix white matter integrity, respectively. Although a small study, the findings will be informative and generate more research investigating this novel approach to treat patients with early AD.
Summary
In this paper, we have explored the possibility that AD be considered a circuit disorder as well as a degenerative disease and that it may be possible to improve or sustain cognitive function in early AD, particularly memory within the integrated cognitive networks involving the hippocampus and cortex, by stimulation at the level of the fornix. This novel strategy supplements our thinking about the etiology of AD and does not discount the prevalent amyloid hypotheses. In fact, the possibility that DBS may be effective suggests that a dual treatment regimen that addresses both amyloid issues and neuronal circuitry for AD patients may be on the horizon.
References
1. Wimo A, Prince M. Alzheimer’s Disease International: World Alzheimer Report 2010: The Global Economic Impact of Dementia. London, United Kingdom: Alzheimer’s Disease International (ADI); 2010.
2. Hardy JA, Selkoe D. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;19;297(5580):353–356.
3. Lyketsos CG, Szekely CA, Mielke MM, et al. Developing new treatments for alzheimer’s disease: The who, what, when, and how of biomarker-guided therapies. Int Psychogeriatr. 2008; 20(5):871–889.
4. Neugroschl J, Sano M. An update on treatment and prevention strategies for alzheimer’s disease. Curr Neurol Neurosci Rep. 2009;9(5):368–376.
5. Golde TE, Schneider LS, Koo EH. Anti-abeta therapeutics in alzheimer’s disease: the need for a paradigm shift. Neuron. 2011;69(2):203–213.
6. Morgan D. Immunotherapy for alzheimer’s disease. J Intern Med. 2011;269(1):54–63.
7. Aisen PS, Saumier D, Briand R, et al. A phase II study targeting amyloid-beta with 3APS in mild-to-moderate alzheimer disease. Neurology. 2006; 67(10):1757–1763.
8. Salloway S, Sperling R, Gilman S, et al. A phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate alzheimer disease. Neurology. 2009; 73(24):2061–2070.
9. Doody RS, Gavrilova SI, Sano M, Dimebon investigators, et al. Effect of dimebon on cognition, activities of daily living, behaviour, and global function in patients with mild-to-moderate alzheimer’s disease: a randomised, double-blind, placebo-controlled study. Lancet. 2008;372(9634):207–215.
10. Green RC, Schneider LS, Amato DA, Tarenflurbil Phase 3 Study Group, et al. Effect of tarenflurbil on cognitive decline and activities of daily living in patients with mild alzheimer disease: a randomized controlled trial. JAMA. 2009;302(23):2557–2564.
11. Holmes C, Boche D, Wilkinson D, et al. Long-term effects of Abeta42 immunisation in Alzheimer’s disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet. 2008;372(9634):216–223.
12. Laxton AW, Tang-Wai DF, McAndrews MP, et al. A phase I trial of deep brain stimulation of memory circuits in Alzheimer’s disease. Ann Neurol. 2010;68:521–534.
13. Sperling RA, Dickerson BC, Pihlajamaki M, et al. Functional alterations in memory networks in early Alzheimer’s disease. Neuromolecular Med. 2010;12(1):27–43.
14. Andrews-Hanna JR, Snyder AZ, Vincent JL, et al. Disruption of large-scale brain systems in advanced aging. Neuron. 2007;56(5):924–935.
15. Sperling RA, Laviolette PS, O’Keefe K, et al. Amyloid deposition is associated with impaired default network function in older persons without dementia. Neuron. 2009;63(2):178–188.
16. Raichle ME, MacLeod AM, Snyder AZ, et al. A default mode of brain function. Proc Natl Acad Sci U S A. 2001;98(2):676–682.
17. Miller SL, Celone K, DePeau K, et al. Age-related memory impairment associated with loss of parietal deactivation but preserved hippocampal activation. Proc Natl Acad Sci U S A. 2008;105(6):2181–2186.
18. Buckner RL, Sepulcre J, Talukdar T, et al. Cortical hubs revealed by intrinsic functional connectivity: Mapping, assessment of stability, and relation to alzheimer’s disease. J Neurosci. 2009;29(6):1860–1873.
19. Seeley WW, Crawford RK, Zhou J, et al. Neurodegenerative diseases target large-scale human brain networks. Neuron. 2009;62(1):42–52.
20. Smith GS, de Leon MJ, George AE, et al. Topography of cross-sectional and longitudinal glucose metabolic deficits in alzheimer’s disease. pathophysiologic implications. Arch Neurol. 1992;49(11):1142–1150.
21. Minoshima S, Giordani B, Berent S, et al. Metabolic reduction in the posterior cingulate cortex in very early alzheimer’s disease. Ann Neurol. 1997 Jul;42(1):85–94.
22. Reiman EM, Caselli RJ, Yun LS, et al. Preclinical evidence of alzheimer’s disease in persons homozygous for the epsilon 4 allele for apolipoprotein E. N Engl J Med. 1996;334(12):752–758.
23. Rabinovici GD, Jagust WJ. Amyloid imaging in aging and dementia: Testing the amyloid hypothesis in vivo. Behaviour Neurol. 2009;21(1):117–128.
24. Buckner RL, Snyder AZ, Shannon BJ, et al. Molecular, structural, and functional characterization of alzheimer’s disease: evidence for a relationship between default activity, amyloid, and memory. J Neurosci. 2005;25(34):7709–7717.
25. Mormino EC, Kluth JT, Madison CM, et al. Episodic memory loss is related to hippocampal-mediated beta-amyloid deposition in elderly subjects. Brain. 2009;132(Pt 5):1310–1323.
26. Klunk WE, Engler H, Nordberg A, et al. Imaging brain amyloid in alzheimer’s disease with pittsburgh compound-B. Ann Neurol. 2004;55(3):306–319.
27. Fripp J, Bourgeat P, Acosta O, et al. Appearance modeling of 11C PiB PET images: characterizing amyloid deposition in alzheimer’s disease, mild cognitive impairment and healthy aging. Neuroimage. 2008; 43(3):430–439.
28. Petersen RC, Jack CR, Jr. Imaging and biomarkers in early alzheimer’s disease and mild cognitive impairment. Clin Pharmacol Ther. 2009;86(4):438–441.
29. Risacher SL, Saykin AJ, West JD, Alzheimer’s Disease Neuroimaging Initiative (ADNI), et al. Baseline MRI predictors of conversion from MCI to probable AD in the ADNI cohort. Curr Alzheimer Res. 2009;6(4):347–361.
30. Powell, TP, Guillery, RW, Cowan WM. A quantitative study of the fornixmamillo-thalamic system. J Anat. 1957;91(4):419–437.
31. Tsivilis D, Vann SD, Denby C, et al. A disproportionate role for the fornix and mammillary bodies in recall versus recognition memory. Nat Neurosci. 2008;11(7):834–842.
32. Wilson CR, Baxter MG, Easton A, Gaffan D. Addition of fornix transection to frontal-temporal disconnection increases the impairment in object-in-place memory in macaque monkeys. Eur J Neurosci. 2008;27(7):1814–1822.
33. Browning PG, Gaffan D, Croxson PL, Baxter MG. Severe scene learning impairment, but intact recognition memory, after cholinergic depletion of inferotemporal cortex followed by fornix transection. Cereb Cortex. 2010;20(2):282–293.
34. Mielke M, Okonkwo O, Oishi K, et al. Fornix integrity and hippocampal volume predict memory decline and progression to AD. Alz Demen. In Press.
35. Mielke MM, Kozauer NA, Chan KC, et al. Regionally-specific diffusion tensor imaging in mild cognitive impairment and alzheimer’s disease. Neuroimage. 2009;46(1):47–55.
36. Bronstein JM, Tagliati M, Alterman RL, et al. Deep brain stimulation for Parkinson disease: an expert consensus and review of key issues. Arch Neurol. 2011;68:165.
37. Sankar T, Tierney TS, Hamani C. Novel applications of deep brain stimulation. Surg Neurol Int. 2012;3:26.
38. Lozano AM. Deep brain stimulation therapy. Br Med J. 2012;344:e1100
39. Moro E, Lang AE, Strafella AP, et al. Bilateral globus pallidus stimulation for huntington’s disease. Ann Neurol. 2004;56(2):290–294.
40. Mayberg HS, Lozano AM, Voon V, et al. Deep brain stimulation for treatment-resistant depression. Neuron. 2005;45(5):651–660.
41. Medtronic. Parkinson’s disease. http://www.medtronic.com/patients/parkinsons-disease/index.htm. Accessed Dec 1, 2012.
42. Oh MY, Seong KH, Abosch A, et al. Long-term hardware-related complications of deep brain stimulation. Neurosurgery. 2002; 50:1268-1276.
43. Toda H, Hamani C, Fawcett AP, et al. The regulation of adult rodent hippocampal neurogenesis by deep brain stimulation. J Neurosurg. 2008;108(1):132–138.
44. Hamani C, Stone SS, Garten A, et al. Memory rescue and enhanced neurogenesis following electrical stimulation of the anterior thalamus in rats treated with corticosterone. Exp Neurol. 2011; 232(1):100–104.
45. Stone SS, Teixeira CM, Devito LM, et al. Stimulation of entorhinal cortex promotes adult neurogenesis and facilitates spatial memory. J Neurosci. 2011;31(38):13469–13484.
46. Laxton AW, Tang-Wai DF, McAndrews MP, et al. A phase I trial of deep brain stimulation of memory circuits in Alzheimer’s disease. Ann Neurol. 2010;68(4):521–534.
47. Alexander GE, Chen K, Pietrini P, et al. Longitudinal PET evaluation of cerebral metabolic decline in dementia: a potential outcome measure in Alzheimer’s disease treatment studies. Am J Psychiatry. 2002;159(5):738–745.
48. Greicius MD, Srivastava G, Reiss AL, Menon V. Default-mode network activity distinguishes alzheimer’s disease from healthy aging: evidence from functional MRI. Proc Natl Acad Sci U S A. 2004;101(13):4637–4642.
49. Buckner RL, Andrews-Hanna JR, Schacter DL. The brain’s default network: Anatomy, function, and relevance to disease. Ann N Y Acad Sci. 2008;1124:1–38.
50. Ito K, Ahadieh S, Corrigan B, Alzheimer’s Disease Working Group, et al. Disease progression meta-analysis model in alzheimer’s disease. Alzheimers Dement. 2010;6(1):39–53.
51. Stern RG, Mohs RC, Davidson M, et al. A longitudinal study of alzheimer’s disease: measurement, rate, and predictors of cognitive deterioration. Am J Psychiatry. 1994;151(3):390–396.




{kind=link}
{kind=link}
{kind=link}
{kind=link}