 by David R. Spiegel, MD; Alexander Pattison, MD; Alexis Lyons, MD; Umer Ansari, MD; Aidan L. Mccroskey, MD; Eric Luehrs MD; Lauren Barr, BS; and Stephanie Le, MD
by David R. Spiegel, MD; Alexander Pattison, MD; Alexis Lyons, MD; Umer Ansari, MD; Aidan L. Mccroskey, MD; Eric Luehrs MD; Lauren Barr, BS; and Stephanie Le, MD
All authors are with the Department of Psychiatry and Behavioral Sciences at Eastern Virginia Medical School in Norfolk, Virginia.
Innov Clin Neurosci. 2017;14(5–6):11–20
Funding: No funding was provided for the preparation of this article.
Financial Disclosures: The authors have no conflicts of interest relevant to the content of this article.
Keywords: Pain, pruritus, nausea, central processing
Abstract Pain, pruritus, and nausea are complex sensory and emotional physiological symptoms that can vary widely between people and even within an individual, depending on the context and meaning of the symptom and the psychological state of the person. This article reviews the acute neural transmission of pain, pruritus, and nausea symptoms, which can begin in the periphery and/or viscera. The subsequent multiple pathways in the central nervous system that become involved in the processing of these symptoms are also discussed. The authors describe human brain imaging studies that have revealed consistent cortical and subcortical networks activated by these symptoms, including sensory, limbic, and associative regions. In particular, the authors discuss information revealed by the studies regarding the primary somatosensory cortex, secondary somatosensory cortex, anterior cingulate cortex, insula, prefrontal cortex and thalamus, are the brain areas most commonly activated by noxious stimuli. Finally, the authors describe treatment options for chronic presentations of these symptoms, which are, in part, based on central nervous processing of these sensations.
Introduction
Heightened somatic awareness (HSA) refers to a greater than average awareness for a variety of physical sensations and symptoms. Individuals with HSA have a tendency to notice and report nonspecific symptoms, such as shortness of breath, presyncope, and a variety of chronic pain conditions, pruritus, and nausea.1 While each sensation has unique neural transmission pathways to the spinal cord, there appears to be a convergent processing of these sensations at the level of the brain itself.
As opposed to acute sensory symptoms, the perpetuation of chronic sensory symptoms has been linked to attributes other than the traditional neural transmission. For instance, HSA is strongly associated with negative affect (i.e., symptoms of depression and anxiety).[1] Between physical symptom reporting and the tendency to experience somatosensory distortion among patients with HSA, the link between HSA and negative effect is consistent with recent neurocognitive theories.[2] Another variable is alterations in neural transmission at the level of the spinal cord (i.e., peripheral and central sensitization).[3] A final variable to consider is alterations in neural transmission at the sensory processing level of the brain, here referred to as the “sensory neuromatrix.” Sensory neuromatrix might be conceptualized as patterns of sensory system activation integrated aberrantly with activity (including emotional, cognitive, and modulatory processes) in brain systems, such as the anterior cingulate cortex, insular cortex, ventrolateral orbitofrontal area, amygdala, striatum, thalamus, hypothalamus, rostral ventromedial medulla, periaqueductal grey, pons (locus coeruleus), red nucleus, and medulla oblongata.[4]
By conceptualizing this process of neural transmissions of acute and chronic pain, pruritus, and nausea within the CNS and by better understanding CNS pathology and its potential effects on chronic sensory symptoms, clinicians might more accurately analyze and more effectively treat chronic sensory symptoms in their patients.
Pain
Pain is a multimodal, complex experience that involves numerous neural sites, including peripheral nerves, the spinal cord, and higher brain centers. The specific receptive properties of thinly myelinated (A-delta fiber) and unmyelinated (C-fiber) nociceptors are determined by membrane-bound transducing ion channel receptors, which are sensitive to temperature, chemical stimuli, and mechanical forces. Upon activation, these ion channel receptors transduce an external stimulus into a change in membrane potential via the opening of a sodium/calcium channel or the closing of a potassium channel (nociceptive transduction or proportionate pain in response to identifiable noxious stimulus).[5]
Nociceptive primary afferents release glutamate, activating postsynaptic glutamate receptors on spinal cord dorsal horn neurons.[6] The second-order neurons cross over at the spinal cord, then travel up the ascending tracts, mainly via the spinothalamic tract to the thalamus. The third-order neurons project to somatosensory cortex and then to prefrontal cortex and amygdala.[7] A confluence of processes, including structural reorganization and sensory gating changes within these circuits, decreases inhibition of these circuits and promotes chronic pain.[8]
One mechanism used to describe the genesis of chronic pain is peripheral sensitization, which constitutes a decreased threshold and increased responsiveness of nociceptors because of post-translational changes in and altered trafficking of transducer receptors and ion channels. This is induced by local inflammatory mediators, and results in pain hypersensitivity symptoms confined to the site of the inflamed tissue. This is referred to as the zone of primary hyperalgesia.[9]
In contrast, chronic neuropathic pain often extends spatially beyond the area of the initially involved root or nerve to create a zone of secondary hyperalgesia, which often becomes independent of the initial noxious event. These symptoms cannot be explained by changes in the peripheral nervous system, but rather reflect changes in spinal and supraspinal networks that culminate in a functional shift of the sensory system from physiological high-threshold nociception to pathological low-threshold pain hypersensitivity.[9]
One major mechanism responsible for this shift of the sensory system is central sensitization, defined broadly as the “increased responsiveness of nociceptive neurons in the CNS to their normal or subthreshold afferent input.” Activity-dependent mechanisms of central sensitization include homosynaptic long-term potentiation (i.e., exaggeration of nociceptor responsiveness) and heterosynaptic potentiation (i.e., recruiting low threshold ABeta fiber inputs into the pain pathway). These mechanisms might be driven and sustained by ectopic activity in the injured nerve.[9]
In addition to the strengthening excitatory synapses in the spinal cord, loss of inhibition by decreasing gamma aminobutryic acid (GABA)-ergic and glycinergic tone also contributes to central hyperexcitability and can be produced by peripheral nerve lesions. Increasing spinal inhibition with intrathecal GABA or by activation of inhibitory interneurons results in an antinociceptive effect, while blocking inhibitory transmission (e.g., by selective ablation of glycinergic dorsal horn interneurons) leads to lowered pain thresholds and the development of hyperalgesia and tactile allodynia.[9]
Pruritus
Chronic pruritus (CPr), or itch lasting six or more weeks, is a major symptom in numerous dermatological and systemic diseases. Similar to chronic pain, CPr (colloquially known as “itchiness”) can have a dramatic impact on the quality of life and can worsen the general condition of the patient considerably.[10]
Itch is described as an unpleasant sensation that evokes the desire to scratch.[11] It is a distinct sensory modality from pain, though both are initiated and mediated by primary sensory neurons with their cell bodies in the dorsal root ganglia (DRG) or trigeminal ganglia. These neurons vary in their somal sizes, expression of ion channels and receptors, regions that they innervate, and electrophysiological properties. Small-diameter DRG neurons with unmyelinated axons (C-fibers) are an important type of neuron for transmitting both pain and pruritus. Despite this similar pathway of neural transmission, pain and itch are perceived differently and cause distinctive behavioral responses. For example, pain stimulus might elicit retraction to avoid tissue injury, while an itch stimulus might instigate scratching to eliminate irritants.[11]
The peripheral pathway of itch is initiated when pruritogens stimulate skin receptors that generate a signaling cascade and action potentials.[12] Two types of itch-sensitive pathways have been identified.[13] These include a histamine-stimulated pathway that uses mechanically insensitive C-fibers (CMi) and a cowhage-stimulated pathway mediated by protease-activated receptors (PAR2) that primarily uses polymodal C-fibers.[13] Just as various C-fibers play a role in pruritus, many different types of receptors can coexist on a single fiber thus enabling primary neurons to function in a polymodal manner.[9,12,14] Certain substances and physical factors, such as temperature, can activate C-fibers and trigger the release of neurotransmitters in the skin via an axon reflex. The itch signal proceeds along spinal nerves’ peripheral processes, spinal ganglia, spinal nerves’ central processes, and spinal cord’s dorsal root and dorsal horn.[11]
After this peripheral pathway, the central pathway of itch begins with a synapse of a primary to secondary afferent neuron in the dorsal horn. The axons of the secondary cells cross over and ascend in the contralateral spinothalamic tract and synapse onto third-order neurons in the thalamus. The axons of the third-order cells then project diffusely to cortical and subcortical regions.[15] Scratching might relieve itch by inhibitory neurons within the spinal cord or by top-down modulation from the supraspinal level.[10] This physiologic pathway can explain how the act of scratching an itch might produce pain in addition to relieving the pruritic sensation, thus illustrating the importance of the neurosensory matrix when evaluating these complaints.
In pathological conditions promoting CPr, sensitization of itch might modify the neural pathway involved, possibly initiating spontaneous itching or inducing hypersensitivity to itch. Sensitization of itch can occur in one or more of the following four ways. First, altered processing of itch by the nervous system can cause nociceptive stimuli that would normally evoke pain to instead evoke itch. Second, peripheral sensitization involves decreased threshold for activation, increased responsiveness, and the presence of ongoing activity of neural pathways. Third, central sensitization involves altered activity of CNS neurons such that stimuli that would normally be nonpruritic become coupled to pruritic sensations.[15] Central sensitization might be caused by altered expression of itch-sensing molecules in the CNS, though the mechanism by which this occurs remains under investigation.[16] Fourth, increased nerve fiber density can also potentially mediate chronic pruriceptive itch.[15,16] It has been postulated that neurotrophins, though not pruritogenic, might contribute to increased nerve fiber density and number. Neurotrophins are a class of growth factors that include nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), neurotrophin 3 (NT-3) and neurotrophin 4/5 (NT-4/5). These molecules bind to three different subtypes of tropomysin-receptor-kinase (Trk) receptors thereby mediating an increase in sensory nerve growth and sprouting.[15]
Nausea
Chronic functional nausea and vomiting include both chronic idiopathic nausea (CIN) and functional vomiting, as defined by the Rome III criteria for functional gastrointestinal (GI) disorders. CIN refers to bothersome nausea occurring at least several times weekly, not necessarily associated with vomiting, with the absence of abnormalities at upper endoscopy or any metabolic disease that explains the nausea. Functional vomiting, likewise, refers to an average of one or more episodes of vomiting weekly in the absence of any eating disorder, rumination, major psychiatric disease, self-induced vomiting, chronic cannabinoid use, CNS abnormalities, or metabolic disease that explains the vomiting. In each of these disorders, criteria need to be fulfilled for the previous three months, with symptom onset at least six months prior to diagnosis.[17]
In order to understand the pathophysiology underlying nausea, it is important to introduce the concept of the dynamic threshold. It is proposed that each individual has a threshold for nausea that changes minute by minute. At any given moment, the threshold depends on the interaction of certain inherent factors of the individual with the more changeable psychological states of anxiety, anticipation, expectation, and adaptation. This dynamic interaction likely explains the inter- and intra-individual variability that is typically encountered in the face of a nauseogenic stimulus.[18]
Briefly, stimuli giving rise to nausea and vomiting activate the chemoreceptor trigger zone (CTZ) in the area postrema and structures deeper in the medulla (“the vomiting center” [VC]) by emetic humoral agents and in the VC by neural input from factors such as vagal afferents and vestibular stimulation and from cerebral structures.[19]
Despite the prevalence and importance of nausea, surprisingly little is known regarding the central mechanisms underlying this sensation. The neurocircuitry involved in emesis has been better characterized. Associated autonomic changes the occur during nausea and emesis (e.g., salivation, sweating) are coordinated at the level of the medulla oblongata. Chemosensitive receptors detect the presence of emetic agents in the blood, and this information is relayed via the area postrema to the nucleus tractus solitarius (NTS). Abdominal vagal afferents that detect gastric tone and contents also project to the NTS. Neurons from the NTS then project to a central-pattern generator, which coordinates the various actions involved in the act of emesis in addition to directly projecting to neurons in the ventral medulla and hypothalamus, from which higher brain areas can be reached.[18]
Pathophysiology of Chronic Pain
In the periphery, after an event that causes direct nerve damage, an inflammatory response ensues. Lysis of inflammatory cells results in the release of various chemical mediators, including serotonin, bradykinin, substance P, histamine, and products from the cyclooxygenase and lipoxygenase pathways of arachidonic acid metabolism. These substances act to sensitize nociceptors in the periphery to subsequent input resulting in increased responsiveness to thermal and mechanical stimuli at the site of injury. This is mediated through thin, unmyelinated C-fiber primary afferent neurons and is referred to as hyperalgesia. Continuous discharge in C fibers might produce sensations of burning pain, whereas intermittent spontaneous discharges in A[d] or A[b] fibers might produce lancinating dysesthesias or paresthesias. This leads to increased susceptibility to firing and increased firing frequency, possibly resulting in not only spontaneous pain, but also central sensitization.20
Under these conditions, central neurons that normally receive high-threshold sensory input might begin to receive input from low-threshold mechanoreceptors, and this information might then be interpreted as nociceptive information ( i.e., allodynia). An alternative hypothesis is that allodynia is caused by a decrease in central inhibition of the mechanically induced nociceptive input. In addition, it is hypothesized that within the spinal cord, collateral sprouting of primary afferent neurons within the spinal cord might occur. According to this model, nerve fibers in deeper laminae that do not normally transmit pain sprout into more superficial regions of the dorsal horn (e.g., laminae I and II) and become receptive to nociceptive input.[21]
Overview of Brain Processing of Sensory Stimuli
The manner in which chronic sensation develops was previously thought to mainly affect the somatosensory system, but now is thought to mainly affect emotional, cognitive, and modulatory areas of the brain. Defining these effects is imperative to understanding how they can modulate these sensations.
Perhaps the most simple effect to understand is somatosensory information, which involves the evaluation of stimulus intensity, (i.e., duration, intensity, frequency) and the somatotopic representation of the stimulus. The cognitive aspects of processing sensory information involves increased “vigilance” toward this sensation and other sensation-related information. Finally, emotional aspects involved with the processing of sensory stimuli include the unpleasant mood that might result from different sensations.[20]
Another negative cognitive and mood effect that impacts pain is catastrophizing. This construct incorporates magnification of pain-related symptoms, rumination about pain, feelings of helplessness, and pessimism about pain-related outcomes, and it is defined as a set of negative emotional and cognitive processes.[22]
Thus, sensorimotor (SM), emotional/affective (EA), cognitive/integrative and modulatory regions of the brain are involved in the complex processing of sensory information, with some areas being involved in more than one domain. Overall, meta-analyses show that chronic sensory experiences produce activation in the primary and secondary somatosensory cortex (SM), insular cortex (SM/EA), anterior cingulate cortex (EA/CI), orbitofrontal cortex (EA/CI) and dorsolateral prefrontal cortex (CI).[20]
Brain Processing of Pain
The rostral anterior cingulate cortex (rACC) connects to the hippocampus, and extends into the periaqueductal grey (PAG) area of the brainstem. It is well known that descending inhibitory pathways in the brain that involve the rACC and PAG play a crucial role in pain modulation.[23] The PAG receives direct projections from regions within the limbic forebrain (i.e., rACC and the amygdala) and can modulate pain perception through brainstem structures (i.e., rostral ventromedial medulla (RVM)), that directly communicate with nociceptive neurons in the dorsal horn of the spinal cord.[23]
The ACC might be divided in subregions that serve different aspects of pain processing. Therefore, one region might be associated with increased or decreased response to pain depending on the exact anatomical location. While primary (S1) and secondary somatosensory cortices (S2) are involved in coding pain stimulus intensity and location, rACC appears to participate in both the affective and attentional concomitants of pain sensation, as well as response selection.[25] Recently published animal studies found augmented synaptic transmission in the ACC in response to long-term exposure to peripheral pain, and there is evidence suggesting that the rACC is required for the reward associated with pain relief.[23] Activation of the rACC in humans has been demonstrated during inhibition of pain, whereas activation of the more posterior parts of the ACC has been associated with increased pain and negative affect in humans.[23]
The role of the hippocampus in pain modulation is likely related to the aversive drive and motivational dimension of pain. Similar to the posited role of the hippocampus, the activation of amygdala in relation to pain is proposed to represent a defensive mechanism that contributes to the recruitment of descending inhibition. Thus, the reduced connectivity between the rACC and hippocampus/amygdala in patients with chronic pain might indicate a weakened defensive response to pain stimuli.[23]
The orbitofrontal cortex (OFC) is involved in sensory integration, reward processing, decision-making, and expectation.[23] A meta-analysis of previously published imaging data revealed that the medial part of the OFC is related to reward whereas the lateral areas are more associated with evaluation of noxious events and motivation to respond.[24] In a recent pain study, the OFC activity was associated with reduction in pain unpleasantness after meditation training, which supports OFC involvement in the processing of incoming pain signals. Moreover the lateral OFC is central for placebo analgesia (compared to opioid analgesia) and for anticipation of pain relief, thus furthering the notion that the OFC plays an essential role in evaluation and inhibition of pain.[24]
Finally, the insular cortex has a primary role in interoception, or the sense of the physiological condition of the body. The interoceptive pathway overlaps with the traditional pain pathways in several areas and additionally relays distinct painful sensory information with homeostatic properties. The parallel and imminent effect is increased pain sensitivity, as the insula is at the core of “the pain matrix” and relays sensory information to the limbic system as well as has a role in autonomic regulation.[26,27]
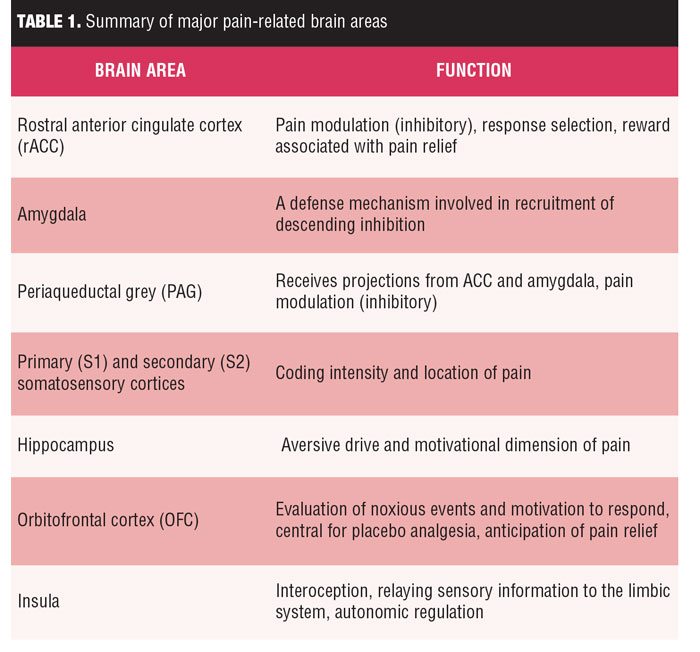 In summary, pain-related brain areas have different patterns of activity with distinct characteristics during the processing of pain. Related to the affective aspects of pain processing, areas such as the insula, inferior frontal gyrus, orbitofrontal cortex, ventrolateral and dorsolateral prefrontal cortex, posterior cingulate cortex (PCC), and ACC show discriminative activity. The S1 and S2 areas as well as the insula are reportedly essential in the sensory processing of pain, with S2 also important for affective processing. Pain processing related to attention has been associated with significantly altered activity in the thalamus, insula, hippocampus, ACC, OFC, dorsolateral prefrontal cortex, and posterior parietal cortex. Finally, it is important to note that among these areas, the insula has been continually reported to be significantly active during the entirety of pain processing. Knowledge of these anatomic areas might be essential for treating chronic pain patients without distinct symptoms, as summarized in Table 1.[28]
In summary, pain-related brain areas have different patterns of activity with distinct characteristics during the processing of pain. Related to the affective aspects of pain processing, areas such as the insula, inferior frontal gyrus, orbitofrontal cortex, ventrolateral and dorsolateral prefrontal cortex, posterior cingulate cortex (PCC), and ACC show discriminative activity. The S1 and S2 areas as well as the insula are reportedly essential in the sensory processing of pain, with S2 also important for affective processing. Pain processing related to attention has been associated with significantly altered activity in the thalamus, insula, hippocampus, ACC, OFC, dorsolateral prefrontal cortex, and posterior parietal cortex. Finally, it is important to note that among these areas, the insula has been continually reported to be significantly active during the entirety of pain processing. Knowledge of these anatomic areas might be essential for treating chronic pain patients without distinct symptoms, as summarized in Table 1.[28]
Brain Processing of Pruritis
The complexity of itch processing in the brain is consistent with the sensation’s multidimensional nature. Cerebral neuropathic pruritus is induced by a cerebral lesion causing subsequent spontaneous firing of damaged neurons. Injury to the area of the brain supplied by the middle cerebral artery, internal capsule, or thalamus—and even the parietal lobe in one patient—has been shown to cause contralateral pruritus.11 Additionally, multiple imaging studies of human brains have shown several cortical regions that are significantly involved in the perception of itch. In these studies, the primary and secondary somatosensory cortices (S1, S2), the insula and anterior cingulate cortex (ACC) and prefrontal cortex (PFC) were frequently activated, commonly in a bilateral manner. Activation of the ACC and insula reflects the affective and emotional elements of the itch experience, with linkage to the limbic system and areas regulating evaluative functions and decision-making, such as the dorsolateral prefrontal cortex (DLPFC).[29,30]
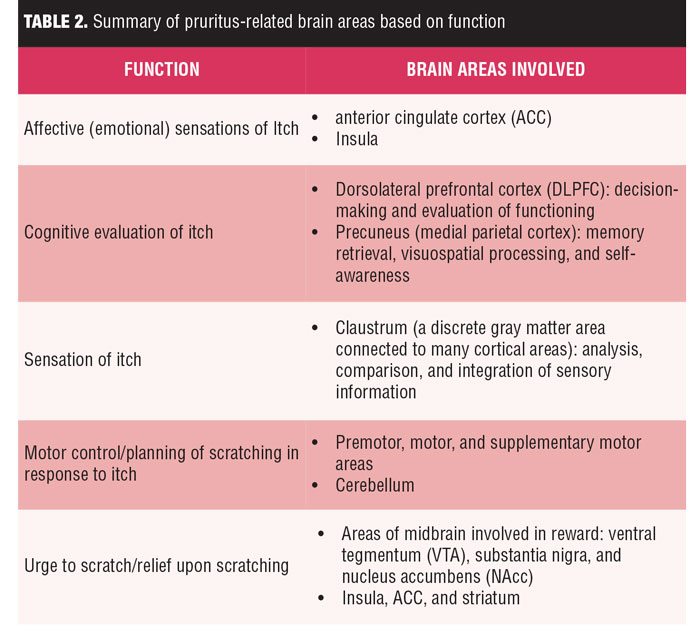 Regions of the brain involved in memory retrieval, visuospatial processing, and self-awareness, such as the precuneus (medial parietal cortex), are activated by the sensation of itch more significantly than during pain processing. The claustrum, a discrete gray matter area connected to many areas of the cortex with a role in the analysis, comparison, and integration of sensory information, has recently been shown to be involved in itch processing. Premotor, motor, and supplementary motor areas as well as the cerebellum are also involved in controlling actual or planned scratching. The activation of both the premotor cortex and supplementary motor area (SMA) areas occurred in most studies, even when the act of scratching itself was prohibited.30 Areas of the midbrain involved in the reward system, such as the ventral tegmentum (VTA), substantia nigra, and nucleus accumbens (NAc), and its connections to the insula, ACC, and striatum might contribute to the urge to scratch and the relief provided by scratching, as summarized in Table 2.[29]
Regions of the brain involved in memory retrieval, visuospatial processing, and self-awareness, such as the precuneus (medial parietal cortex), are activated by the sensation of itch more significantly than during pain processing. The claustrum, a discrete gray matter area connected to many areas of the cortex with a role in the analysis, comparison, and integration of sensory information, has recently been shown to be involved in itch processing. Premotor, motor, and supplementary motor areas as well as the cerebellum are also involved in controlling actual or planned scratching. The activation of both the premotor cortex and supplementary motor area (SMA) areas occurred in most studies, even when the act of scratching itself was prohibited.30 Areas of the midbrain involved in the reward system, such as the ventral tegmentum (VTA), substantia nigra, and nucleus accumbens (NAc), and its connections to the insula, ACC, and striatum might contribute to the urge to scratch and the relief provided by scratching, as summarized in Table 2.[29]
Brain Processing of Nausea
Once nausea is recognized, sustained brain activation is demonstrated in a broader network of interoceptive (e.g., stomach awareness), limbic, somatosensory, and cognitive processing brain areas. A correlation analysis demonstrated that subjects who had greater anterior insula activation on neuroimaging following transition to strong nausea also demonstrated greater activation in midcingulate cortex.[31] This finding suggests a closer linkage between these specific regions within the brain circuitry supporting the perception of nausea. These results are consistent with the characterization of nausea as a multidimensional perceptual state encompassing interoceptive, emotional, and cognitive domains.[31]
GI sensation elicited by inflated balloon distention has facilitated mapping of visceral interoceptive circuits, which most consistently show activation of the anterior and posterior insula, as well as midcingulate cortex. However, it should be noted that visceral balloon inflation stimuli typically produce pain sensations instead of nausea. Thus, while interoception from the esophagus and stomach likely play an important role in the perception of nausea, such afferent signals might be necessary but are not sufficient to produce nausea.[31]
Previous studies have reported that higher-level stimuli (e.g., visual scenes of vomiting) could induce emotional disgust and nausea via activation of the anterior insula and midcingulate cortex. These structures have also been implicated in salience detection, which relates to assigning homeostatic relevance for both internal and external sensory inputs to the brain. Hence, we can further postulate that nausea emerges from activation in a broad brain network that includes salience-processing regions, which alter interoceptive signaling in order to alert the suffering individual to enact the appropriate autonomic/motor response.[31]
Increasing sustained activation common to affective/emotional circuitries was also noted in perigenual (pg) ACC, OFC, NAcc, and VTA. These structures likely support the aversive nature of nausea, and the OFC in particular might attribute hedonic valence to interoceptive afference. The pgACC is an important subregion of the ACC that is also strongly related to emotion.[31]
Brain activity in bilateral prefrontal cortical regions (PFC), especially dorsolateral, demonstrated sustained postrating activation with increasing nausea levels. These prefrontal cortical regions might support the cognitive/evaluative disruption.[31]
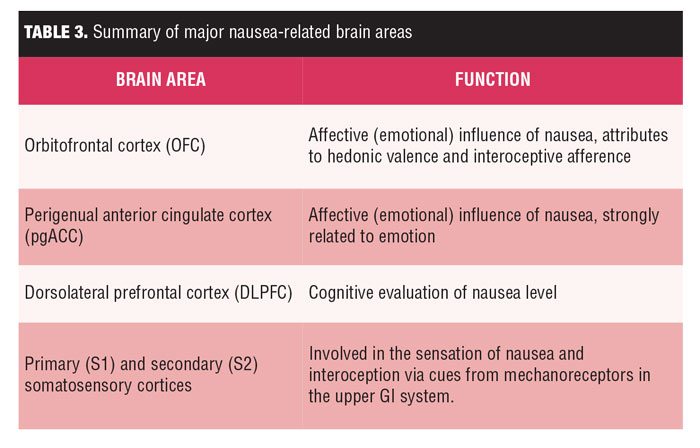 Both S1 and S2 also demonstrated sustained postrating activation proportionate to increasing nausea sensation. Visceral balloon inflation studies elicit activation in both interoceptive and somatosensory brain regions.[31] S1 activity has been reported to be localized to a somatotopic location (ventrolateral subregion of the postcentral gyrus) consistent with the upper GI cortical representation reported in animals and humans.[31] Interestingly, lesion studies suggest that somatosensory cortices might also support interoception, and for nausea, might relate to cues from mechanoreceptors overlying the epigastrium (e.g., gastric tachyarrhythmia is increased during nausea, as summarized in Table 3.[31]
Both S1 and S2 also demonstrated sustained postrating activation proportionate to increasing nausea sensation. Visceral balloon inflation studies elicit activation in both interoceptive and somatosensory brain regions.[31] S1 activity has been reported to be localized to a somatotopic location (ventrolateral subregion of the postcentral gyrus) consistent with the upper GI cortical representation reported in animals and humans.[31] Interestingly, lesion studies suggest that somatosensory cortices might also support interoception, and for nausea, might relate to cues from mechanoreceptors overlying the epigastrium (e.g., gastric tachyarrhythmia is increased during nausea, as summarized in Table 3.[31]
Functional Somatic Symptoms
Patients with functional somatic symptoms (FSS) experience alterations of attention, anticipation, and pain memories. A poorly elucidated network of higher brain functions, previously called the “pain matrix,” is now referred to as the “neuromatrix.” The former was a misnomer, as the neuromatrix is active in various conscious processes, not just pain.[32]
The insula, which shares interactions with the ACC, is another important area implicated in FSS. The insula is known to modulate pain processing and paradigms when painful stimulation is expected. Activation of the insula has been reported primarily for cutaneous pain rather than visceral pain. In patients with fibromyalgia, experiments on pain expectation demonstrated that in patients with nondermatomal somatosensory deficits, the posterior part of the insula showed hypometabolic activation.[32[
The lateral nociceptive system, which includes the insula, indicated consistent hyperperfusion, whereas the ACC and the more affective motivational system did not. The increased activation in the insula might be correlated with the exaggerated expectancy of pain and attention in FSS patients. This might be correlated with the anterior part of the insula, which is associated with cognitive-affective aspects of pain. In functional somatic syndrome patients, the increased insular activation is correlated with heightened sensory coding of stimuli that are innocuously coded in healthy patients. This effect has also been shown in patients with irritable bowel syndrome.[32] Medial and posterior parts of the insula are thought to be involved in these somatosensory discriminative abilities. It is hypothesized that an increased interoception is at work and is mediated via missing inhibitory descending prefrontal input and/or continued ascending arousal.[32]
The lamina I spinothalamic pathway allows for the integration of visceral-somatic information within the brainstem, thalamus, and cortex. Visceral-somatic information converges cortically within the ACC and insula. The ACC has been classically subdivided into a subgenual/pregenual affective component and a dorsal ACC/anterior middle cingulate cortex (aMCC) cognitive component. Nociceptive studies across visceral-somatic stimuli elicit robust cingulate gyrus activations; visceral stimuli frequently activate pregenual and dorsal ACC, whereas noxious cutaneous stimuli more robustly activate the MCC. The dorsal ACC/aMCC is uniquely positioned for integration and is where affectively ladened information, visceral-somatic processing, motivated behavior, and cognitive control converge. Shackman, et al recently proposed this region as a critical integrator of negative affect, pain, and cognitive control.[33]
Suggested Theoretical Neural Framework for HAS-Type Illness
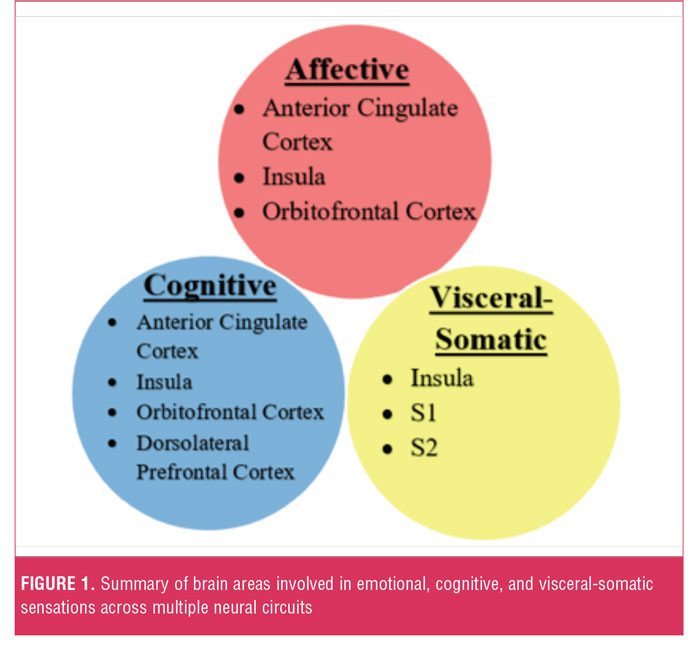 Aberrant circuit interactions across neural systems mediating visceral-somatic perception, emotional processing/ awareness, and cognitive control serve critical roles in the neurobiology of somatosensory amplification. Cortical-subcortical-brainstem-spinal cord interactions are theorized to mediate the amplification of visceral-somatic sensations. Thus, the somatosensory cortices (S1 and S2) and insult cortex encode sensory information, such as the location and duration of pain, nausea, or itch.[34] Important cognitive processes in somatosensory amplification include negative expectation bias (ACC, OFC, insula, hippocampal formation, and brainstem); negative attentional bias (ACC, amygdala, dorsolateral prefrontal cortex); and pain catastrophizing (ACC, dorsolateral prefrontal cortex, insula). Affective processes linked to somatosensory amplification include alexithymia (ACC, insula, and amygdala) and dysphoric-anxious mood (ACC, Insula, OFC) (Figure 1).[33]
Aberrant circuit interactions across neural systems mediating visceral-somatic perception, emotional processing/ awareness, and cognitive control serve critical roles in the neurobiology of somatosensory amplification. Cortical-subcortical-brainstem-spinal cord interactions are theorized to mediate the amplification of visceral-somatic sensations. Thus, the somatosensory cortices (S1 and S2) and insult cortex encode sensory information, such as the location and duration of pain, nausea, or itch.[34] Important cognitive processes in somatosensory amplification include negative expectation bias (ACC, OFC, insula, hippocampal formation, and brainstem); negative attentional bias (ACC, amygdala, dorsolateral prefrontal cortex); and pain catastrophizing (ACC, dorsolateral prefrontal cortex, insula). Affective processes linked to somatosensory amplification include alexithymia (ACC, insula, and amygdala) and dysphoric-anxious mood (ACC, Insula, OFC) (Figure 1).[33]
Clinical correlations. While a comprehensive review of specific disorders of heightened somatic awareness are beyond the scope of this manuscript, three representative disorders of HSA with pain, itch, and nausea sensations (fibromyalgia, atopic dermatitis, and idiopathic gastroparesis, respectively) are summarized in Table 4.[31,34–36,40–49]
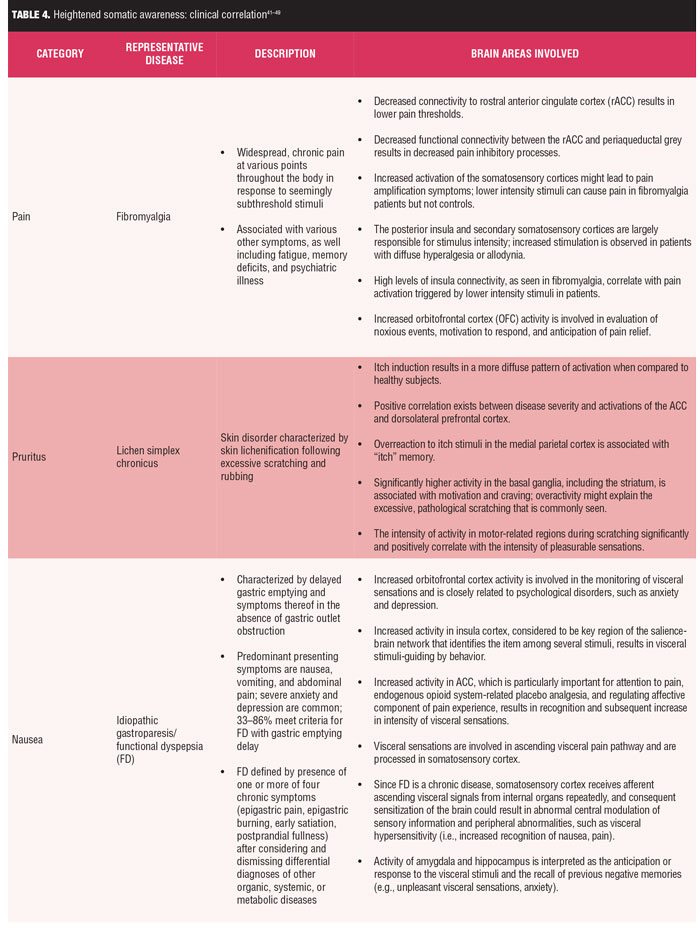
Fibromyalgia is a condition in which there is an imbalance of the functional connectivity within the pain network and shows strengthened connectivity of relevant brain regions involved in pain processing as well as significantly reduced connectivity of areas involved in pain inhibitory modulation.[34]
Relying on published consensus statements, gastroparesis is defined by the presence of dyspeptic symptoms and the documented delay in gastric emptying of ingested nutrients in the absence of gastric outlet obstruction. Traditionally, gastroparesis was thought to be characterized by anorexia and postprandial symptoms with nausea, vomiting, bloating, early satiation, and fullness. Pain was not considered to be typical and raised the question of functional dyspepsia, although it is often present with significant nausea.[35] Regardless, strong nausea has been shown to result in sustained activation in a broad network of interoceptive, limbic, somatosensory, and cognitive processing brain areas, which results in the accompanying unpleasant effect and increased attention to the sensation of nausea.[31]
Finally, atopic dermatitis (AD) is an inflammatory skin condition of intense itch and scratching. Functional magnetic resonance imaging (MRI) shows a positive correlation between disease severity and activations of the anterior cingulate cortex. The latter is likely to be associated with cognition/evaluation of itch stimuli and/or the urge to scratch. Additionally, there is significantly higher activity in the basal ganglia, including the striatum, for patients with Alzheimer’s disease (AD). Activation in the basal ganglia is associated with motivation and craving. An overactivity in this region could possibly explain the excessive, pathological scratching commonly seen in AD patients. Finally, the intensity of activity in motor-related regions during scratching is significantly and positively correlated with the intensity of pleasurable sensations in AD patients. This significant positive correlation suggests that scratching-induced pleasurability might augment activity in motor-related regions, irrespective of whether the skin was itchy or not.[36]
Thus, while each of the above disorders present with different primary symptoms, they all share HSA. Imaging studies in each of the disorders have found functional abnormalities in overlapping and unique brain areas associated with sensory-related processing.[34–36]
Treatment Implications
A full review of the treatment for these chronic sensations is beyond the scope of this review; however, while each of the aforementioned sensations has sensory specific treatments, two classes of neuromodulatory medications have been shown to effectively treat the central processing aberrations that might occur in chronic pain, nausea, and pruritus symptoms: anticonvulsants and antidepressants. More specifically, those anticonvulsants selectively bind to the alpha2–delta subunit of neuronal voltage-gated calcium channels (i.e., gabapentin and pregabalin), resulting in inhibition of calcium currents, thus decreasing the excitatory transmitter release and spinal sensitization.[37] Additionally, those antidepressants that interfere in the neuronal reuptake of neurotransmitters such as serotonin and norepinephrine (i.e., tricyclic antidepressants, mirtazapine, duloxetine) result in recruitment of noradrenergic-descending pathways as well as the peripheral recruitment of noradrenaline from sympathetic fibers sprouting into dorsal root ganglia. There is evidence of definitive treatment successes in all three of these chronic sensory sensations with these classes of medications.[37–40]
References
- Schrepf A, Harper DE, Williams DA, et al. Somatic awareness and tender points in a community sample. J Pain. 2016;0(0):170–176. doi:10.1016/j.jpain.2016.08.009.
- Brown RJ, Skehan D, Chapman A, et al. Physical symptom reporting is associated with a tendency to experience somatosensory distortion. Psychosom Med. 2012;74(6):648–655. doi:10.1097/PSY.0b013e3182595358.
- Oh H-M, Chung M. Botulinum toxin for neuropathic pain: a review of the literature. Toxins (Basel). 2015;7(8):3127–3154. doi:10.3390/toxins7083127.
- Borsook D. Neurological diseases and pain. Brain. 2012;135(Pt 2):320–344. doi:10.1093/brain/awr271.
- Vardeh D, Mannion RJ, Woolf CJ, et al. Toward a mechanism-based approach to pain diagnosis. J Pain. 2016;17(9 Suppl):T50–69. doi:10.1016/j.jpain.2016.03.001.
- Bardoni R. Role of presynaptic glutamate receptors in pain transmission at the spinal cord level. Curr Neuropharmacol. 2013;11(5):477–483. doi:10.2174/1570159X11311050002.
- Fong A, Schug SA. Pathophysiology of pain. Plast Reconstr Surg. 2014;134:8S–14S. doi:10.1097/PRS.0000000000000682.
- Scioli-Salter ER, Forman DE, Otis JD, et al. The shared neuroanatomy and neurobiology of comorbid chronic pain and PTSD. Clin J Pain. 2015;31(4):363–374. doi:10.1097/AJP.0000000000000115.
- Ikoma A, Cevikbas F, Kempkes C, Steinhoff M. Anatomy and neurophysiology of pruritus. Semin Cutan Med Surg. 2011;30(2):64–70. doi:10.1016/j.sder.2011.04.001.
- Mochizuki H, Kakigi R, Adam R, et al. Central mechanisms of itch. Clin Neurophysiol. 2015;126(9):1650–1660. doi:10.1016/j.clinph.2014.11.019.
- Wallengren J. Neuroanatomy and neurophysiology of itch. Dermatol Ther. 2005;18(4):292–303. doi:10.1111/j.1529-8019.2005.00041.x.
- Jovanovic M. Current concepts of pathophysiology, epidemiology and classification of pruritus. Srp Arh Celok Lek. 2014;142(1–2):106–112. doi:10.2298/SARH1402106J.
- Dhand A, Aminoff MJ. The neurology of itch. Brain. 2014;137(2):313–322.
- Metz M, Ständer S. Chronic pruritus—pathogenesis, clinical aspects and treatment. J Eur Acad Dermatology Venereol. 2010;24(11):1249–1260. doi:10.1111/j.1468-3083.2010.03850.x.
- Potenzieri C, Undem BJ. Basic mechanisms of itch. Clin Exp Allergy. 2012;42(1):8–19. doi:10.1111/j.1365-2222.2011.03791.x.
- Han L, Dong X. Itch mechanisms and circuits. Annu Rev Biophys. 2014;43:331–355. doi:10.1146/annurev-biophys-051013-022826.
- Patel A, Sayuk GS, Kushnir VM, Gyawali CP. Sensory neuromodulators in functional nausea and vomiting: predictors of response. Postgrad Med J. 2013;89(1049):131–136. doi:10.1136/postgradmedj-2012-131284.
- Singh P, Yoon SS, Kuo B. Nausea: a review of pathophysiology and therapeutics. Therap Adv Gastroenterol. 2016;9(1):98–112. doi:10.1177/1756283X15618131.
- Törnblom H, Abrahamsson H. Chronic nausea and vomiting: insights into underlying mechanisms. Neurogastroenterol Motil. 2016;28(5):613–619. doi:10.1111/nmo.12837.
- Borsook D, Sava S, Becerra L. The pain imaging revolution: advancing pain into the 21st century. Neuroscientist. 2010;16(2):171–185. doi:10.1177/1073858409349902.
- Stacey BR. Management of peripheral neuropathic pain. Am J Phys Med Rehabil. 2005;84(3 Suppl):S4–16.
- Tracey I, Mantyh PW. The cerebral signature for pain perception and its modulation. Neuron. 2007;55(3):377–391. doi:10.1016/j.neuron.2007.07.012.
- Jensen KB, Loitoile R, Kosek E, et al. Patients with fibromyalgia display less functional connectivity in the brain’s pain inhibitory network. Mol Pain. 2012;8:32. doi:10.1186/1744-8069-8-32.
- Kringelbach M. The functional neuroanatomy of the human orbitofrontal cortex: evidence from neuroimaging and neuropsychology. Prog Neurobiol. 2004;72:341–372.
- Peyron R, Laurent B, García-Larrea L. Functional imaging of brain responses to pain: a review and meta-analysis (2000). Neurophysiol Clin Neurophysiol. 2000;30(5):263–288. doi:10.1016/S0987-7053(00)00227-6.
- Karshikoff B, Jensen KB, Kosek E, et al. Why sickness hurts: a central mechanism for pain induced by peripheral inflammation. Brain Behav Immun. 2016;57:38–46. doi:10.1016/j.bbi.2016.04.001.
- Craig AD. How do you feel? interoception: the sense of the physiological condition of the body. Nat Rev Neurosci. 2002;3(8):655–666. doi:10.1038/nrn894.
- Kang DH, Son JH, Kim YC, et al. Neuroimaging studies of chronic pain. Korean J Pain. 2010;23(3):159. doi:10.3344/kjp.2010.23.3.159.
- Yosipovitch G, Mochizuki H. Neuroimaging of Itch as a Tool of Assessment of Chronic Itch and Its Management. Springer Berlin Heidelberg; 2015:57–70. doi:10.1007/978-3-662-44605-8_4.
- Mochizuki H, Papoiu A, Yosipovitch G. Brain processing of itch and scratching. In: Carstens E, Akiyama T (eds). Itch: Mechanisms and Treatment. Boca Raton, Florida: CRC Press/Taylor & Francis; 2014:391–409.
- Napadow V, Sheehan JD, Kim J, et al. The brain circuitry underlying the temporal evolution of nausea in humans. Cereb Cortex. 2013;23(4):806–813. doi:10.1093/cercor/bhs073.
- Boeckle M, Schrimpf M, Liegl G, Pieh C. Neural correlates of somatoform disorders from a meta-analytic perspective on neuroimaging studies. NeuroImage Clin. 2016;11:606–613. doi:10.1016/j.nicl.2016.04.001.
- Perez DL, Barsky AJ, Vago DR, et al. A neural circuit framework for somatosensory amplification in somatoform disorders. J Neuropsychiatry Clin Neurosci. 2015;27(1):e40–50. doi:10.1176/appi.neuropsych. 13070170.
- Cifre I, Sitges C, Fraiman D, et al. Disrupted functional connectivity of the pain network in Ffbromyalgia. Psychosom Med. 2012;74(1):55–62. doi:10.1097/PSY.0b013e3182408f04.
- Bielefeldt K. Gastroparesis: concepts, controversies, and challenges. Scientifica (Cairo). 2012;2012:1–19. doi:10.6064/2012/424802.
- Mochizuki H, Schut C, Nattkemper LA, Yosipovitch G. Brain mechanism of itch in atopic dermatitis and its possible alteration through noninvasive treatments. Allergol Int. 2017;66(1):14–21. doi:10.1016/j.alit.2016.08.013.
- Ständer S, Weisshaar E, Luger TA. Neurophysiological and neurochemical basis of modern pruritus treatment. Exp Dermatol. 2008;17(3):161–169. doi:10.1111/j.1600-0625.2007.00664.x.
- Hasler WL. Newest drugs for chronic unexplained nausea and vomiting. Curr Treat Options Gastroenterol. October 2016:1–15. doi:10.1007/s11938-016-0110-2.
- Khouzam HR. Psychopharmacology of chronic pain: a focus on antidepressants and atypical antipsychotics. Postgrad Med. 2016;128(3):323–330. doi:10.1080/00325481.2016.1147925.
- Kremer M, Salvat E, Muller A, et al. Antidepressants and gabapentinoids in neuropathic pain: mechanistic insights. Neuroscience. July 2016. doi:10.1016/j.neuroscience. 2016.06.057.
- Schmidt-Wilcke T, Ichesco E, Hampson JP, et al. Resting state connectivity correlates with drug and placebo response in fibromyalgia patients. NeuroImage Clin. 2014;6:252–261. doi:10.1016/j.nicl.2014.09.007.
- Wager TD, Atlas LY, Lindquist MA, et al An fMRI-based neurologic signature of physical pain. N Engl J Med. 2013;368(15):1388–1397. doi:10.1056/NEJMoa1204471.
- Segerdahl AR, Mezue M, Okell TW, et al. The dorsal posterior insula subserves a fundamental role in human pain. Nat Neurosci. 2015;18(4):499–500. doi:10.1038/nn.3969.
- Lotti T, Buggiani G, Prignano F. Prurigo nodularis and lichen simplex chronicus. Dermatol Ther. 2008;21(1):42–46. doi:10.1111/j.1529-8019.2008.00168.x.
- Papoiu ADP, Nattkemper LA, Sanders KM, et al. Brain’s reward circuits mediate itch relief. a functional MRI study of active scratching. PLoS One. 2013;8(12):e82389. doi:10.1371/journal.pone.0082389.
- Mochizuki H, Tanaka S, Morita T, et al. The cerebral representation of scratching-induced pleasantness. J Neurophysiol. 2014;111(3):488–498. doi:10.1152/jn.00374.2013.
- Mochizuki H, Papoiu ADP, Nattkemper LA, et al. Scratching induce overactivity in motor-related regions and reward system in chronic itch patients. J Invest Dermatol. 2015;135(11):2814–2823. doi:10.1038/jid.2015.223.
- Kovacic K, Miranda A, Chelimsky G, et al. Chronic idiopathic nausea of childhood. J Pediatr. 2014;164(5):1104–1109. doi:10.1016/j.jpeds.2014.01.046.
- Kovacic K, Di Lorenzo C. Functional nausea in children. J Pediatr Gastroenterol Nutr. 2016;62(3):365–371. doi:10.1097/MPG.0000000000001076.




{kind=link}
{kind=link}
{kind=link}
{kind=link}