 by Vaibhav Rastogi, MBBS; Lauren L. Donnangelo; Ganesh Asaithambi, MD; Sharatchandra Bidari, MD; Anna Y. Khanna, MD; and Vishnumurthy Shushrutha Hedna, MD
by Vaibhav Rastogi, MBBS; Lauren L. Donnangelo; Ganesh Asaithambi, MD; Sharatchandra Bidari, MD; Anna Y. Khanna, MD; and Vishnumurthy Shushrutha Hedna, MD
Drs. Rastogi, Asaithambi, Khanna, and Hedna and Ms. Donnangelo are from the Department of Neurology and Dr. Bidari is from the Department of Radiology—All from the University of Florida College of Medicine, Gainesville, Florida.
Innov Clin Neurosci. 2015;12(5–6):20–26.
Funding: No funding was provided for the preparation of this article.
Financial disclosures: The authors have no conflicts of interest relevant to the content of this article.
Key words: Cerebral amyloid angiopathy, lobar intracerebral hemorrhage, recurrence, inflammation, corticosteroid
Abstract: Background: Recurrent lobar intracerebral hemorrhage is more commonly associated with cerebral amyloid angiopathy and less likely associated with hypertension. Cerebral amyloid angiopathy-related inflammation is a subgroup of cerebral amyloid angiopathy that can present with lobar intracerebral hemorrhage, encephalopathy, and seizures; wherein corticosteroids may facilitate favorable outcome. Whether recurrence of lobar intracerebral hemorrhage in cerebral amyloid angiopathy is related to cerebral amyloid angiopathy-related inflammation is unknown. Case presentation: A 68-year-old woman presented with an acute onset of confusion. She was known to have a history of recurrent lobar intracerebral hemorrhage related to cerebral amyloid angiopathy. Brain imaging revealed previous sequelae of cerebral amyloid angiopathy and a new lobar intracerebral hemorrhage. An empirical diagnosis of cerebral amyloid angiopathy-related inflammation was made given the patent’s clinical course of recurrence. Utilizing current evidence of criteria used to diagnose cerebral amyloid angiopathy-related inflammation, corticosteroid therapy was initiated with significant improvement in clinical and imaging characteristics. Discussion: Inflammatory pathways incited as a result of cerebrovascular amyloid deposition play a vital role in pathogenesis of cerebral amyloid angiopathy-related inflammation. We highlight the need to consider corticosteroid therapy in patients presenting with recurrent lobar intracerebral hemorrhage in the setting of cerebral amyloid angiopathy since inflammation may play a role in its pathophysiology. Evidence in the literature is sparse to suggest that cerebral amyloid angiopathy-related inflammation might be the root cause for the lobar intracerebral hemorrhage recurrence in cerebral amyloid angiopathy. Further studies are needed to identify mechanisms of recurrent hemorrhage, its correlations with cerebral amyloid angiopathy-related inflammation, and the potential role of corticosteroid therapy.
Introduction
Cerebral amyloid angiopathy (CAA) is a progressive age-related vasculopathy due to excess deposition of beta-amyloid (Ab) in leptomeningeal and cortical arterioles. Mainly found in the elderly and demented patients, CAA presents with hemorrhagic stroke, ischemic stroke, cognitive impairment, and transient neurological symptoms.[1] Population-based autopsy studies associate CAA in 55 to 59 percent of demented elderly,[2] 90 percent of Alzheimer’s patients,[3,4] and 28 to 38 percent of nondemented elderly cohort.[2] CAA-related intracerebral hemorrhage (ICH) can manifest as lobar macrohemorrhages, cortical microhemorrhages, convexal subarachnoid hemorrhage, or superficial siderosis on imaging studies.[1] The lobar hemorrhages involve predominantly cortical and subcortical regions5 with a predilection to temporal and occipital lobes6 but can involve multiple sites.[7]
Recurrence in ICH has been reported up to 24 percent[8] in cases of primary ICH cases and majority of them are lobar hemorrhages related to CAA.[9,10] SMASH-U criteria (structural lesion, medication, amyloid angiopathy, systemic/other disease, hypertension, undetermined), a pathogenetic classification proposed for the etiologic classification of ICH, also highlights the importance of CAA in the causation of ICH.[11] At present, there is no established treatment regimen to curb the recurrence of CAA-related lobar ICH.[12]
CAA- related inflammation (CAA-RI) is a lesser known entity in which corticosteroid therapy has shown some promise.[13] CAA-RI and its relation with recurrence of lobar ICH in CAA is largely unexplored. Here, we report a patient with recurrent CAA-related lobar ICH and inflammation who responded to corticosteroid therapy and developed improvement in higher cortical functioning and significant reduction in lesion severity on magnetic resonance imaging (MRI).
Case Presentation
A 68-year-old woman with a history of hypertension presented to the hospital with acute confusion in June 2013. Her exam was consistent with limb kinetic apraxia of the left arm and alien limb phenomenon of the left upper extremity and left arm and leg clumsiness. Brain imaging revealed a right frontal ICH with associated surrounding vasogenic edema and extension into the local subarachnoid space. There were innumerable microhemorrhages bilaterally, compatible with amyloid angiopathy. Her presentation was attributed to CAA and her home therapy was adjusted and she was discharged. A few months later, in April 2014, she was readmitted with acute onset confusion, speech difficulty and transient left side weakness. History was also suggestive of some transient seizure-like activity as per family history. Examination confirmed transcortical motor aphasia and mild left hemiparesis. Brain imaging revealed a new left frontal lobar ICH and was also evident was encephalomalacia in the right frontal lobe from previous ICH. Multifocal remote infarcts and scattered punctate foci of restricted diffusion in the inferior left cerebellum and the bilateral frontal and parietal lobes, corresponding to amyloid, were also noticed. She was treated conservatively with blood pressure control and an anticonvulsant regimen.
During her admission in May 2014, she was admitted again with acute confusion, headache, and vomiting and was hospitalized for workup of her acute encephalopathy. MRI brain showed a new right temporo-parietal ICH with surrounding vasogenic edema. On initial neurologic exam, the patient exhibited impaired orientation, comprehension, repetition, and recall. A mild left facial droop was also noted. No further abnormalities in mental status, cranial nerve function, strength, or sensation were detected. Biochemical findings were within normal limits except for elevated single-stranded DNA IgG antibody at 83 EU and C-reactive protein at 13mg/L. MRI images were obtained on admission (Figure 1).
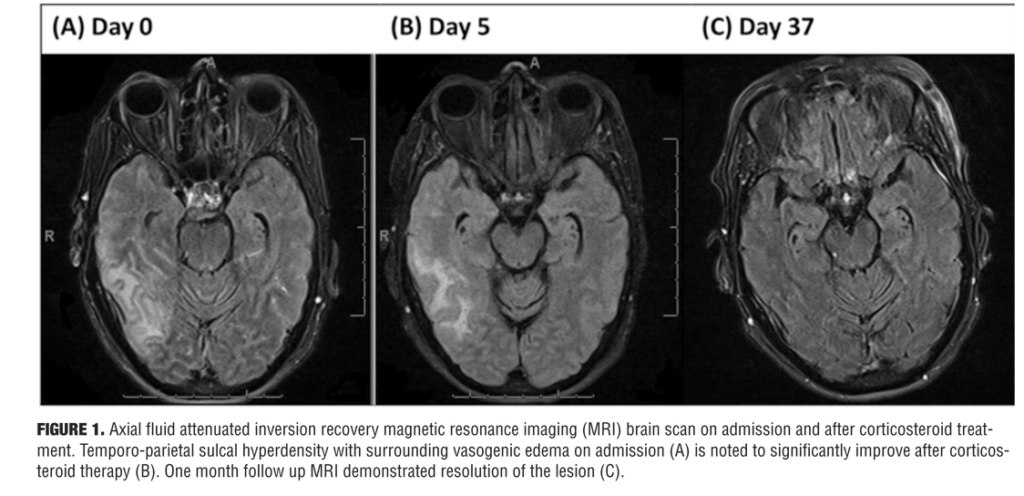
Overall, the patient’s presentation on admission was highly suggestive of encephalopathy related to her CAA. The patient’s family declined further spinal tap and leptomeningeal biopsy. Given her multiple recurrence of lobar hemorrhage, we decided to initiate corticosteroid therapy entertaining the diagnosis of CAA-RI. A short course of corticosteroid pulse therapy was initiated with daily 1g of methylprednisolone sodium succinate administered intravenously for five days. Five consecutive days into steroid initiation, the patient completed repeat brain MRI and Montreal Cognitive Assessment (MoCA) examination. Her MoCA scores demonstrated significant improvement from 7 on Day 1 to 14 on Day 3 (Figure 2). There was accompanying radiological improvement noted on the brain MRI (Figure 1). The patient was discharged on 40mg daily oral prednisone and 75mg daily oral azathioprine. At one-month follow-up, MRI scans showed almost complete resolution of the lesion (Figure 1), and her MoCA score had improved to 19. She continues to show progress to date with the MoCA score reaching 27 at six months follow-up.

Discussion
CAA commonly involves small cortical arteries and rarely veins.[14] Ab deposition results in thickened vasculature, smooth muscle destruction, endothelial dysfunction,[15] cerebral dysautoregulation, ongoing inflammation, and disruption of the blood-brain barrier.[16] The apolipoprotein E (Apo E, particularly the e4 allele) gene located on chromosome 19 is considered the linkage between Alzheimer’s disease and CAA.17 CAA-RI is also termed cerebral amyloid angiitis, primary angiitis of CNS associated with CAA, and Ab-related angiitis. CAA-RI can present with progressive encephalopathy, seizures, headache, and focal neurological deficits.[18] CAA-RI in comparison to CAA has few distinctive features, including younger age at diagnosis, decreased prevalence of cognitive and neurologic deficits, leptomeningeal enhancement on contrast imaging, and better response to corticosteroid therapy.[19] The relatively younger age, recurrence of ICH, presenting symptoms, and radiographic findings prompted the diagnosis of CAA-RI in our case.
CAA-RI pathophysiology presumably stems from the immune response to the cerebral Ab deposits. The support for this theory comes from immunomodulation studies where vaccine administration in Alzheimer’s disease subjects led to the development of a clinical picture of acute to sub-acute meningoencephalitis, similar to CAA-RI and autoantibodies to Ab42 amyloid form.[20] Also, increased levels of autoantibodies against Ab40 and Ab42 have been noted in the cerebrospinal fluid21 and against Ab42 in the serum of patients with CAA-RI.[22] Ab deposits even result in partial activation of CD4 cells in close vicinity of major histocompatibility complex class II antigens that are expressed by macrophages after engulfing Ab.[23] CAA-RI has two pathological subtypes that may or may not co-exist: a perivasculitic form wherein there is visible perivascular multinuclear giant cells infiltration in the brain parenchyma and a vasculitic form (or transmural granulomatous vasculitis) where direct invasion of the vessel wall by inflammatory mediators are seen.[24]
CAA-RI is a distinct entity that complicates CAA, and no data so far exist to suggest CAA-RI plays a role in CAA related ICH. In CAA-RI, there is Ab deposition leading to various immunomodulation pathway activations, including the release of inflammatory mediator-triggering of complement pathway[25,26] and matrix metalloproteinase-9 activation.[27] These deposits can also incite ion channel formation, toxicity of the cell, disruption of the blood-brain barrier permeability,[25] and proliferation and activation of the monocyte/macrophage.[28] These inflammatory pathways ultimately cause disruption of the cerebrovasculature,[29] which sheds light on the key role of inflammation in CAA-related ICH. Various precipitating factors, including advanced age, hypertension, Alzheimer’s disease, use of antithrombotics, minor trauma, and anti-amyloid therapy such as bapineuzumab, play roles in the development of CAA-related ICH.[30]
Since Apo E gene e4 allele is strongly associated with CAA-related ICH,[17] CAA-RI,[3] early onset CAA related ICH, and early recurrence of ICH,[32] we postulate that the e4 allele might be the epicenter with higher genetic predilection of immune response to Ab in these individuals, linking all of these manifestations. From the above, we further postulate that CAA-RI may be involved in the CAA-related ICH development especially in cases of recurrence. Further research is required to decipher our theory of genetic linkage between CAA-related lobar ICH and CAA-RI.
Brain imaging and biochemical testing play central roles in the diagnosis of CAA-RI. Biochemical analysis involves estimation of cerebrospinal fluid concentrations of Ab.[33] Danve et al[13] noted that inflammatory markers, such as erythrocyte sedimentation rate and C-reactive protein, were elevated in as many as 30 percent of patients with CAA-RI[13] and were also elevated in our patient. Given the patient’s reluctance for further spinal taps, we did not evaluate the cerebrospinal fluid concentrations of Ab and autoantibodies against Ab40 and Ab42, these studies may be an important step in attaining the diagnosis of CAA-RI. Genetic testing for Apo E gene, especially e2 and e4 alleles, may be considered in patients with Alzheimer’s or of older age in order to ascertain their risks for CAA-related ICH and CAA-RI. Key imaging characteristics on brain MRI that correlate to CAA include lobar cerebral microhemorrhages[34] with a predisposition to parietal lobes[6] and leukoaraiosis[35] (deep cerebral white matter imaging changes due to axonal destruction), mild gliosis, and demyelination.[36] Other relevant MRI changes are localized convexity subarachnoid hemorrhage, cortical superficial siderosis (hemosiderin accumulation in superficial cerebral cortical layers),[37] and silent acute ischemic lesion (especially on diffusion weighed imaging).[38] When there is high clinical suspicion of CAA-RI but radiological findings are not definitive, brain biopsy may prove useful.[39]
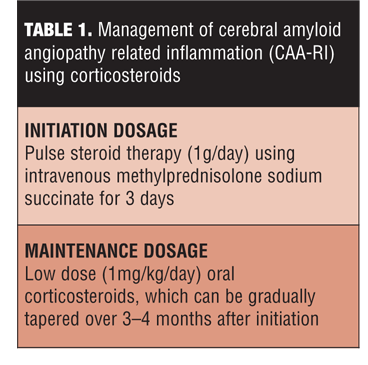
CAA-RI treatment as highlighted by multiple studies is primarily focused on corticosteroid therapy.[39,40] High dose (1g/day) of intravenous methylprednisolone for three days might be a reasonable choice at initiation. The remission can be maintained using a maintenance regimen of low-dose oral corticosteroids (approximately 1mg/kg/day), which can be gradually tapered off over 3 to 4 months (Table 1).[18,39] Studies have shown approximately 1 to 3 weeks are required from treatment initiation to observe the response to treatment.[31]
The exact mechanism of action of corticosteroids in CAA-RI is not known. An in-vitro study by Previti et al[41] has demonstrated that corticosteroids can decrease the pro-inflammatory effects of Ab deposition by increasing interleukin-6 and matrixmetalloproteinase-2 levels in the late inflammatory phase within vascular smooth muscle cells. Another plausible theory is the potential decrease in the surrounding area of vasogenic edema induced by the corticosteroids.[40] The mitigation of immune-related inflammation might be another mechanism for corticosteroid response as demonstrated by the normalization of the increased levels of autoantibodies against Ab40 and Ab42 at three months after initiation of corticosteroids.[21,22] The beneficial effects of immunosuppressants can also be explained by the tendency of CAA-RI to relapse in drug-free periods.[18] Few studies have indicated that perivasculitic forms have the most optimistic prognosis[42] and better response to corticosteroid treatment. This might be due to presence of more vasogenic edema in association with perivasculitic form as compared to the vasculitic form, which has more cytotoxic involvement.[40]
Other immunosuppressive agents employed in the treatment of CAA-RI include cyclophosphamide, azathioprine, methotrexate, and mycophenolate mofetil, but further studies are needed to determine their true level of benefit.[13] Cyclophosphamide may be an alternative of corticosteroid therapy when administered as 100mg/day for the initiation dose for nine months and 50mg/day as the maintenance dose.[43] Methotrexate 20mg/week, in addition to corticosteroids, can also benefit the patient.[44] Azathioprine 75mg/day is another effective alternative for the maintenance corticosteroid therapy.[45] We also noticed absence of recurrence on addition of azathioprine to corticosteroids as a part of the maintenance regimen in our patient.
CAA-related lobar ICH has a high tendency for recurrence.[7] This recurrence can be attributed to the inflammatory variant of CAA. Hoshi et al[46] reported a case with recurrent CAA-related ICH that was treated with corticosteroids; they noted an increase in serum anti-nuclear antibody titers and erythrocyte sedimentation rate, which suggests an inflammatory process. From our clinical observation, we also believe that there might be an increase in recurrence associated with CAA-RI as compared to CAA alone; our patient presented with multiple recurrences of CAA-related ICH that terminated after corticosteroid initiation. CAA-RI treated with immunosuppressants is associated with a good prognosis; approximately 73 percent of patients treated with immunosuppressants have shown good outcomes, with Modified Rankin Scale scores of 3 or lower.[19] Studies have observed complete remission in a majority of patients that have lasted up to five years.[13]
Conclusion
CAA-RI may be an underlying etiology for recurrent CAA-related lobar ICH. Lobar ICH is an integral component of Boston criteria[47,48] for CAA diagnosis, and inflammation might be underlying pathogenesis for its recurrence. Thus, we propose the addition of recurring lobar ICH to the criteria suggested by Chung et al18 for probable CAA-RI (Table 2).

Corticosteroid therapy is the treatment of choice for CAA-RI; it might also be effective to prevent CAA-related lobar ICH recurrence and should be started as soon as the diagnosis of probable CAA-RI is made. In-depth studies are warranted in order to establish the genetic and pathophysiologic correlation between CAA-RI and CAA-related lobar ICH. Randomized controlled trials are necessary to evaluate the efficacy of immunosuppressive therapies, including corticosteroids, in CAA-RI as a form of prevention in recurring ICH among CAA patients.
Author Contributions
VR and LLD drafted the manuscript. LLD administered the MOCA examinations. GA, SB, and AYK revised the paper critically for intellectual content. VSH conceived the study and participated in its design and coordination. All authors read and approved the final manuscript.
Acknowledgments
We would like to extend our thanks to the patient and her family for allowing us to carry out this work and to all who participated in her interdisciplinary care.
References
1. Yamada M, Naiki H. Cerebral amyloid angiopathy. Prog Mol Biol Transl Sci. 2012;107:41–78.
2. Keage HA, Carare RO, Friedland RP, et al. Population studies of sporadic cerebral amyloid angiopathy and dementia: a systematic review. BMC Neurol. 2009;9:3.
3. Kalaria RN, Ballard C. Overlap between pathology of Alzheimer disease and vascular dementia. Alzheimer Dis Assoc Disord. 1999;13 Suppl 3:S115–123.
4. Jellinger KA. Alzheimer disease and cerebrovascular pathology: an update. J Neural Transm. 2002;109(5-6):813–836.
5. Samarasekera N, Smith C, Al-Shahi Salman R. The association between cerebral amyloid angiopathy and intracerebral haemorrhage: systematic review and meta-analysis. J Neurol Neurosurg Psychiatry. 2012;83(3):275–281.
6. Rosand J, Muzikansky A, Kumar A, et al. Spatial clustering of hemorrhages in probable cerebral amyloid angiopathy. Ann Neurol. 2005;58(3):459–462.
7. Hirohata M, Yoshita M, Ishida C, et al. Clinical features of non-hypertensive lobar intracerebral hemorrhage related to cerebral amyloid angiopathy. Eur J Neurol. 2010;17(6):823–839.
8. Passero S, Burgalassi L, D’Andrea P, Battistini N. Recurrence of bleeding in patients with primary intracerebral hemorrhage. Stroke. 1995;26(7):1189–1192.
9. Hill MD, Silver FL, Austin PC, Tu JV. Rate of stroke recurrence in patients with primary intracerebral hemorrhage. Stroke. 2000;31(1):123–127.
10. Neau JP, Ingrand P, Couderq C, et al. Recurrent intracerebral hemorrhage. Neurology. 1997;49(1):106–113.
11. Meretoja A, Strbian D, Putaala J, et al. SMASH-U: a proposal for etiologic classification of intracerebral hemorrhage. Stroke. 2012;43(10):2592–2597.
12. Yeh SJ, Tang SC, Tsai LK, Jeng JS. Pathogenetical subtypes of recurrent intracerebral hemorrhage: Designations by SMASH-U classification system. Stroke. 2014;45(9):2636–2642.
13. Danve A, Grafe M, Deodhar A. Amyloid beta-related angiitis: a case report and comprehensive review of literature of 94 cases. Semin Arthritis Rheum. 2014;44(1):86–92.
14. Thal DR, Ghebremedhin E, Rüb U, et al. Two types of sporadic cerebral amyloid angiopathy. J Neuropathol Exp Neurol. 2002;61(3):282–293.
15. Attems J, Jellinger K, Thal DR, Van Nostrand W. Review: sporadic cerebral amyloid angiopathy. Neuropathol Appl Neurobiol. 2011;37(1):75–93.
16. Smith EE, Greenberg SM. Beta-amyloid, blood vessels, and brain function. Stroke. 2009;40(7):2601–2606.
17. Greenberg SM, Rebeck GW, Vonsattel JP, et al. Apolipoprotein E epsilon 4 and cerebral hemorrhage associated with amyloid angiopathy. Ann Neurol. 1995;38(2):254–259.
18. Chung KK, Anderson NE, Hutchinson D, et al. Cerebral amyloid angiopathy related inflammation: three case reports and a review. J Neurol Neurosurg Psychiatry. 2011;82(1):20–26.
19. Salvarani C, Hunder GG, Morris JM, et al. Ab-related angiitis: comparison with CAA without inflammation and primary CNS vasculitis. Neurology. 2013;81(18):1596–1603.
20. Orgogozo JM, Gilman S, Dartigues JF, et al. Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology. 2003;61(1):46–54.
21. DiFrancesco JC, Brioschi M, Brighina L, et al. Anti-Ab autoantibodies in the CSF of a patient with CAA-related inflammation: a case report. Neurology. 2011;76(9):842–844.
22. Hermann DM, Keyvani K, van de Nes J, et al. Brain-reactive b-amyloid antibodies in primary CNS angiitis with cerebral amyloid angiopathy. Neurology. 2011;77(5):503–505.
23. Melzer N, Harder A, Gross CC, et al. CD4(+) T cells predominate in cerebrospinal fluid and leptomeningeal and parenchymal infiltrates in cerebral amyloid b-related angiitis. Arch Neurol. 2012;69(6):773–777.
24. McHugh JC, Ryan AM, Lynch T, et al. Steroid-responsive recurrent encephalopathy in a patient with cerebral amyloid angiopathy. Cerebrovasc Dis. 2007;23(1):66–69.
25. Revesz T, Holton JL, Lashley T, et al. Genetics and molecular pathogenesis of sporadic and hereditary cerebral amyloid angiopathies. Acta Neuropathol. 2009;118(1):115–130.
26. Ghiso J, Rostagno A, Tomidokoro Y, et al. Genetic alterations of the BRI2 gene: familial British and Danish dementias. Brain Pathol. 2006;16(1):71–79.
27. Lee JM, Yin K, Hsin I, et al. Matrix metalloproteinase-9 in cerebral-amyloid-angiopathy-related hemorrhage. J Neurol Sci. 2005;229-230:249–254.
28. Yamada M, Itoh Y, Shintaku M, et al. Immune reactions associated with cerebral amyloid angiopathy. Stroke. 1996;27(7):1155–1162.
29. Yamada M. Brain hemorrhages in cerebral amyloid angiopathy. Semin Thromb Hemost. 2013;39(8):955–962.
30. Yamada M. Predicting cerebral amyloid angiopathy-related intracerebral hemorrhages and other cerebrovascular disorders in Alzheimer’s disease. Front Neurol. 2012;3:64.
31. Kinnecom C, Lev MH, Wendell L, et al. Course of cerebral amyloid angiopathy-related inflammation. Neurology. 2007;68(17):1411–1416.
32. O’Donnell HC, Rosand J, Knudsen KA, et al. Apolipoprotein E genotype and the risk of recurrent lobar intracerebral hemorrhage. N Engl J Med. 2000;342(4):240–245.
33. Hernandez-Guillamon M, Delgado P, Penalba A, et al. Plasma b-amyloid levels in cerebral amyloid angiopathy-associated hemorrhagic stroke. Neurodegener Dis. 2012;10(1–4):320–323.
34. Greenberg SM, Vernooij MW, Cordonnier C, et al. Cerebral microbleeds: a guide to detection and interpretation. Lancet Neurol. 2009;8(2):165–174.
35. Debette S, Markus HS. The clinical importance of white matter hyperintensities on brain magnetic resonance imaging: systematic review and meta-analysis. BMJ. 2010;341:c3666.
36. Pantoni L, Garcia JH. Pathogenesis of leukoaraiosis: a review. Stroke. 1997;28(3):652–659.
37. Linn J, Herms J, Dichgans M, et al. Subarachnoid hemosiderosis and superficial cortical hemosiderosis in cerebral amyloid angiopathy. AJNR Am J Neuroradiol. 2008;29(1):184–186.
38. Cadavid D, Mena H, Koeller K, Frommelt RA. Cerebral beta amyloid angiopathy is a risk factor for cerebral ischemic infarction. A case control study in human brain biopsies. J Neuropathol Exp Neurol. 2000;59(9):768–773.
39. Sakaguchi H, Ueda A, Kosaka T, et al. Cerebral amyloid angiopathy-related inflammation presenting with steroid-responsive higher brain dysfunction: case report and review of the literature. J Neuroinflammation. 2011;8:116.
40. Kloppenborg RP, Richard E, Sprengers ME, et al. Steroid responsive encephalopathy in cerebral amyloid angiopathy: a case report and review of evidence for immunosuppressive treatment. J Neuroinflammation. 2010;7:18.
41. Previti ML, Zhang W, Van Nostrand WE. Dexamethasone diminishes the pro-inflammatory and cytotoxic effects of amyloid beta-protein in cerebrovascular smooth muscle cells. J Neuroinflammation. 2006;3:18.
42. Schwab S, Steiner T, Aschoff A, et al. Early hemicraniectomy in patients with complete middle cerebral artery infarction. Stroke. 1998;29(9):1888–1893.
43. Fountain NB, Lopes MB. Control of primary angiitis of the CNS associated with cerebral amyloid angiopathy by cyclophosphamide alone. Neurology. 1999;52(3):660–662.
44. Wong SH, Robbins PD, Knuckey NW, Kermode AG. Cerebral amyloid angiopathy presenting with vasculitic pathology. J Clin Neurosci. 2006;13(2):291–294.
45. Luppe S, Betmouni S, Scolding N, Wilkins A. Cerebral amyloid angiopathy related vasculitis: successful treatment with azathioprine. J Neurol. 2010;257(12):2103–2105.
46. Hoshi K, Yoshida K, Nakamura A, et al. Cessation of cerebral hemorrhage recurrence associated with corticosteroid treatment in a patient with cerebral amyloid angiopathy. Amyloid. 2000;7(4):284–288.
47. Greenberg SM, Briggs ME, Hyman BT, et al. Apolipoprotein E epsilon 4 is associated with the presence and earlier onset of hemorrhage in cerebral amyloid angiopathy. Stroke. 1996;27(8):1333–1337.
48. Knudsen KA, Rosand J, Karluk D, Greenberg SM. Clinical diagnosis of cerebral amyloid angiopathy: validation of the Boston criteria. Neurology. 2001;56(4):537–539.




{kind=link}
{kind=link}
{kind=link}
{kind=link}