
 by David R. Spiegel, MD; Aidan L. McCroskey, MD; and Branden A. Deyerle, MD
by David R. Spiegel, MD; Aidan L. McCroskey, MD; and Branden A. Deyerle, MD
All with the Department of Psychiatry and Behavioral Sciences, Eastern Virginia Medical School, Norfolk, Virginia.
Innov Clin Neurosci. 2016;13(3–4):32–41.
Funding: No funding was received for the preparation of this manuscript.
Financial disclosures: The authors have no conflicts of interest relevant to the content of this article.
Key words: Transient global amnesia, hippocampus, neurobiology, delusions
Abstract: Transient global amnesia is a clinical syndrome characterized by the sudden onset of anterograde amnesia, accompanied by repetitive questioning, sometimes with a retrograde component, lasting up to 24 hours, without compromise of other neurologic function. Neuroimaging after an acute transient global amnesia event often shows transient perturbation of specific hippocampal circuits that are involved in memory processing. Critical clinical distinctions, such as between transient global amnesia and other forms of transient amnesic episodes, as well as important clues to the underlying pathophysiologies are herein reviewed. Finally, we discuss the role of hippocampal insufficiency in the neurobiology of delusions.
Introduction
Transient amnesic syndromes are clinical phenomena that occur commonly in acute medical settings. Results from imaging and epidemiological studies have recently revealed distinctions that enable differentiation between various transient amnesic syndromes as well as important clues to their underlying pathophysiologies.
Transient global amnesia (TGA) is a clinical syndrome that was established by Hodges and Warlow in 1990.[1] Criteria are listed in Table 1.

Neuroimaging studies performed after an episode of TGA display transient alteration of the specific hippocampal circuits that contribute to the creation and storage of memories.[2] Episodes last for a mean duration of approximately six hours. The incidence of TGA is approximately 2.9 to 10 people per 100,000 worldwide.[3] It typically affects patients between 50 and 80 years of age, at an average age of 61. Anterograde amnesia gradually diminishes within the course of a few hours and patients return to their baseline status, though they may retain a dense amnesic gap for the duration of the episode.
Proposed causes for TGA include ischemia,[4,5] migraine,[3,6–8] epileptic seizure,[8] venous congestion,[9] and emotional arousal or psychological disturbances.[10] The most widely accepted mechanism of TGA involves cerebrovascular disease of the thromboembolic type.[3]
This article presents the differential diagnosis in a patient with transient amnesia and a history of vascular disease treated with multiple surgical interventions as well as a history of anxiety. Discussion of an ischemia-mediated hypothesis for the pathogenesis of TGA and insight into the neurobiology of delusion follows.
Case Report
Our patient was a 75-year-old woman with a past medical history significant for peripheral arterial/vascular disease with atherosclerosis and ischemia for which she took aspirin, clopidogrel, and simvastatin; atrial fibrillation managed with sotalol; stage two chronic kidney disease for which she took furosemide; hypertension managed with lisinopril; and anxiety managed with citalopram 20mg/day. She presented with symptoms of very short distance claudication in the left lower extremity and pain at rest in the left foot.
She had undergone multiple endovascular interventions in the past. And about six weeks prior to this hospitalization, she underwent repeat percutaneous transluminal balloon angioplasty of the left superficial femoral artery and popliteal artery. She also is status/post left femoral-popliteal artery bypass and femoral-tibial artery bypass surgery and percutaneous transluminal angioplasties.
During this hospitalization, diagnostic aortography revealed left external iliac artery stenosis and left superficial femoral artery stent occlusion with reconstitution of the popliteal artery below the knee. Our patient was admitted for a repeat iliofemoral endarterectomy and femoral to below-knee popliteal bypass.
On the day of planned surgery, the third day of her hospital admission, nurses noticed that the patient had developed an “altered mental status (AMS).” The patient became paranoid, stating to a nurse, “You guys are trying to pull something over me.” Nursing notified the vascular surgeon, who decided to put the surgery on hold. Prior to this point, the patient’s sleep-wake cycle had been normal. The patient refused to allow assessment of vital signs or completion of physical examination, blood draw, radiologic studies, and treatments including heparin and oxygen administration. When the vascular surgeon reiterated that the consequences of not receiving treatment could include loss of a limb, the patient responded, “Then I’ll lose it and you’ll have your consequences.” The patient voiced paranoid thoughts “that everyone was trying to convince her that she was crazy and take her blood.”
At that point, we were consulted to evaluate the patient’s AMS and her decision-making capacity. On initial evaluation, she exhibited inappropriate laughter and cursing. She refused the Vigilant A section of the Confusion Assessment Method-Intensive Care Unit11 and received a zero on the Richmond Agitation Sedation Scale score. Cognitively, she was oriented times 2, and gave an unusual answer regarding year, i.e., “the year of ammeninity.” Her attention span remained intact, and she was otherwise alert. The patient refused to undergo further cognitive evaluation. She could not describe what was medically wrong with her, other than stating, “People think I am crazy.” Her thought process was goal directed and thought content significant for paranoid delusions, as she refused surgery on the grounds that she did not trust the surgeons and therefore wanted to leave. Since her decision-making was based on delusional ideation, she was determined to lack decision-making capacity to refuse surgery. We recommended intramuscular aripiprazole, which our patient also refused.
Approximately 5.5 hours after the onset of the patient’s change in behavior, she suddenly became cooperative and allowed hospital staff to proceed with her evaluation and treatment. On review of laboratory results, the patient’s hepatic function panel returned within normal limits with the exception of a slightly low total protein and slightly elevated aspartate aminotransferase. Urinalysis tests were negative for infection, demonstrating only trace leukocyte esterase. Gamma-glutamyl transpeptidase, vitamin B12, folic acid, and thiamine levels were within normal ranges. Troponin level did not suggest an acute myocardial infarction.
Chest x-ray was negative for infection. Cardiac nonstress testing revealed normal left ventricular systolic function with an ejection fraction over 75 percent. Electrocardiogram (EKG) revealed sinus rhythm.
Electroencephalography (EEG) found no seizure activity, although it did reveal diffuse background slowing. Computed tomography (CT) scan of the head revealed only mild cortical atrophy, with neither hippocampal atrophy nor enlargement of the lateral or third ventricles identified. Carotid peripheral vascular laboratory (PVL) identified moderate stenosis of 50 to 69 percent in the bilateral internal carotid arteries associated with atherosclerotic plaque. Transcranial PVL showed evidence of hemodynamically significant stenosis in the basilar artery.
The day after this episode, the patient was more cooperative, or viewed alternatively, no longer had paranoid delusions. In fact, our patient had no memory of either her paranoid delusions or its impact on her behavior. The Vigilant A section of the Confusion Assessment Method-Intensive Care Unit11 was intact, and the Richmond Agitation Sedation Scale score was zero. Cognitively, she was oriented times 3, without fluctuation of mentation, and without auditory or visual hallucinations.
On the second day after the episode, the Vigilant A section of the Confusion Assessment Method-Intensive Care Unit11 was again intact, and she again received a zero on the Richmond Agitation Sedation Scale score. The patient was alert, aware, and attentive, without auditory or visual hallucinations or delusions. She scored a 30/30 on the Mini-Mental Status Examination (MMSE), and seemed to have returned to her baseline.
Discussion
Differential diagnoses. TGA is a benign, self-limited selective disturbance in anterograde memory that occurs in middle-aged and elderly individuals, consistent with our patient’s demographic. The differential diagnosis for TGA includes several other transient amnesic disorders with varying epidemiology, course of illness, associated neuropsychiatric aspects, and work-up (Tables 2 and 3).[1,3–10,13–15,21,59–66] These include transient epileptic amnesia, transient ischemic attack, and partial complex seizures.

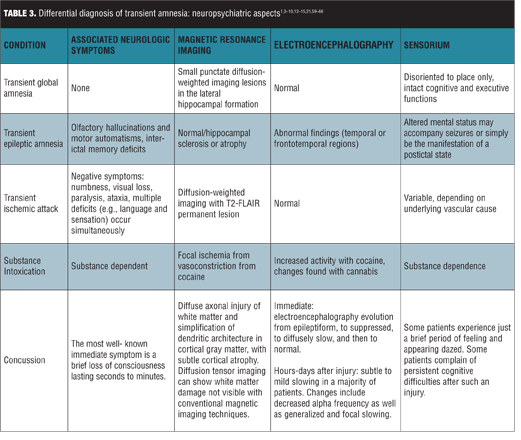 Transient epileptic amnesia (TEA). TEA is a rare form of temporal lobe epilepsy characterized by focal seizures manifesting as recurrent transient episodes of isolated memory loss. TEA differs from partial complex seizures, inasmuch as the latter typically presents with more neurological symptoms, including aura, decreased awareness of the event, automatisms, and postictal state. The postictal state is usually responsible for the amnestic episode of partial complex seizures, whereas the primary feature of TEA is isolated memory loss.12 In contrast to features that favor a diagnosis of epilepsy including a brief duration,[3,13] occurrence on awakening,[13] and multiple attacks,[14] our patient’s episode persisted for approximately 5.5 hours, began in the middle of the day, and presented as a single event. She did not demonstrate motor automatisms often present in TEA.[14] Her episode included the anxiety and repetitive questioning more likely featured in TGA than in amnesic seizures,[16] with agitation, apprehension, and paranoia. Her episode involved anterograde amnesia as opposed to the more prominent retrograde amnesia found in TEA.[17] Furthermore, EEG did not demonstrate ictal activity, which reduced our suspicion for TEA, though a normal EEG or nonspecific EEG abnormalities do not necessarily exclude the diagnosis.[18]
Transient epileptic amnesia (TEA). TEA is a rare form of temporal lobe epilepsy characterized by focal seizures manifesting as recurrent transient episodes of isolated memory loss. TEA differs from partial complex seizures, inasmuch as the latter typically presents with more neurological symptoms, including aura, decreased awareness of the event, automatisms, and postictal state. The postictal state is usually responsible for the amnestic episode of partial complex seizures, whereas the primary feature of TEA is isolated memory loss.12 In contrast to features that favor a diagnosis of epilepsy including a brief duration,[3,13] occurrence on awakening,[13] and multiple attacks,[14] our patient’s episode persisted for approximately 5.5 hours, began in the middle of the day, and presented as a single event. She did not demonstrate motor automatisms often present in TEA.[14] Her episode included the anxiety and repetitive questioning more likely featured in TGA than in amnesic seizures,[16] with agitation, apprehension, and paranoia. Her episode involved anterograde amnesia as opposed to the more prominent retrograde amnesia found in TEA.[17] Furthermore, EEG did not demonstrate ictal activity, which reduced our suspicion for TEA, though a normal EEG or nonspecific EEG abnormalities do not necessarily exclude the diagnosis.[18]
Transient ischemic attack (TIA). Isolated amnesia without other focal neurologic deficits, though rare, can manifest during a TIA or stroke. And the presence of vascular risk factors increases concern for this diagnosis. Transient amnesia may be the sole or primary manifestation of unilateral hemispheric stroke limited to the hippocampus or thalamus; in these cases, however, brain CT or magnetic resonance imaging (MRI) would reveal lesions in limbic structures that account for the memory deficit.[4] In contrast, in patients with TGA, CT scans of the brain are usually normal or may show incidental findings, and additional focal neurological deficits are likely to be present during the attack.[3] Our patient lacked CT scan findings indicative of stroke, TIA, or any focal neurological deficits, and her amnestic episode lasted over five hours, exceeding the typical timeframe of TIA of less than one hour.[19]
Brain MRI with diffusion-weighted imaging (DWI) is performed in most cases to exclude TIA. All pathologies affecting the hippocampus are associated with distinct lesion patterns on MRI, and especially DWI has the ability to demonstrate even minute and transient hippocampal lesions.[20] Most patients with TGA have small punctate DWI lesions in the lateral hippocampal formation.[21] Stroke affecting the hippocampus generates patterns according to vascular pathology. These lesions may be complete, dorsal, lateral, or small and circumscribed, and additional lesions may be present elsewhere in the territory of the PCA. DWI demonstrates ischemic abnormalities in about half of clinically defined TIA patients that may vanish or persist despite transient clinical symptoms.[22] One limitation of our patient’s evaluation was that it did not include an MRI with DWI, which can provide a means to further confirm the etiology of amnesia.
Delirium. While common in a hospital setting in older patients, individuals with delirium classically demonstrate a more global confusion and inattention. On gross evaluation, our patient did not demonstrate the decreased attention span and waxing and waning type of mentation characteristic of delirium. Unfortunately, at that time, we were unable to perform a full CAM-ICU or MMSE, thus it is not definitive that no other cognitive impairment was present. This is a limitation in the patient’s assessment, especially when discussing the differential diagnosis of TGA and delirium. However, she did exhibit paranoid delusions, (e.g., she believed her care team was attempting to cause her harm).
Psychogenic amnesia. Psychogenic amnesia may also be confused with TGA. These patients differ from those with TGA in that memory for their personal life histories is much more severely affected than is their ability to learn and retain new information; that is, they have isolated retrograde amnesia.[36] Our patient retained knowledge of autobiographical data and demonstrated anterograde amnesia, making psychogenic amnesia unlikely.
Arterial or venous ischemia. Though TGA patients have fewer vascular risk factors than TIA patients, the prevalence of vascular risk factors in TGA patients exceeds that found in healthy controls.[37] The etiology of TGA could be explained by an ischemic event due to either arterial ischemia (atherosclerotic or thromboembolic) or venous ischemia.[37,38] Anxiety causes hyperventilation, which results in vasoconstriction, and may potentially exacerbate these ischemic processes.[39] This may explain the high prevalence of comorbid emotional distress[3,8] and anxious personality traits in TGA patients, including our patient. Valsalva-like activities (which block venous return through the superior vena cava, blocking well-oxygenated blood from entering the sensitive hippocampus) are common triggers of TGA onset. Insufficient jugular-vein valves (which cause brief retrograde transmission of high venous pressure to cerebral veins, also blocking well-oxygenated blood from entering the sensitive hippocampus) are also common in TGA patients.[40,41] The CA1 neurons present in the hippocampus, which are thought to be the structural and functional correlate of TGA, exhibit selective vulnerability to metabolic stress.[42] Stress may be one trigger involved in the emergence of transient lesions in the hippocampal CA1 region.[43] Therefore, temporary hypoperfusion per the aforementioned ischemia could explain why memory structures are affected first.
Other possible organic diagnoses. Other possibilities included in the differential diagnosis for TGA include head injury,[8] anoxic event,[23] hypoglycemia,[24] drug or alcohol intoxication,[25–27] drug or alcohol withdrawal, herpes simplex encephalitis,[28] and other causes of toxic-metabolic encephalopathy and delirium.[29] Our patient reported no history of trauma, and CT failed to demonstrate evidence of head injury or temporal lobe changes indicative of herpes simplex encephalitis. She had not experienced an anoxic event such as respiratory failure, and her lab studies, including serum glucose testing, were unremarkable. She denied use of drugs or alcohol, and demonstrated no signs or symptoms of intoxication or withdrawal. Her lack of alcohol use is also relevant since it makes a diagnosis of Wernicke’s encephalopathy less likely. Though Wernicke’s encephalopathy may present primarily with amnesia,[30] our patient’s normal thiamine levels make Wernicke’s encephalopathy an unlikely cause for her episode. Discontinuation of her opiates was also unlikely the cause of resolution of the patient’s abnormal behavior on the planned day of surgery, as opiates typically do not result in psychoses.
Drug-drug interaction. Our patient was taking aspirin, clopidogrel, simvastatin, sotalol, furosemide, linsinopril, and citalopram at the time of her amnestic episode. While there are no drug-drug interactions among these medications known to result in memory loss, the possibility of a drug-drug interaction cannot be ruled out. It has been reported that polypharmacy can result in substantial drug-drug interactions.31
Polypharmacy and a lack of knowledge about complex drug interactions can result in unexpected side effects, as the literature regarding drug-drug interactions focuses almost exclusively on paired combinations.[31] On a more general level, benzodiazepines and non-benzodiazepine hypnotics have been associated with transient memory loss. Although our patient was not taking any of these medications before her amnestic event, the use of these medications needs to be considered in the evaluation of transient global amnesia.[32] Additionally, statins as a class have been reported to cause memory loss; our patient was taking simvastatin and we cannot rule this out as a possible contributor to her memory loss.[33] Furthermore, while serotonin-norepinephrine reuptake inhibitors have been associated with decreased long-term potentiation in the hippocampus, selective serotonin reuptake inhibitors, including citalopram, have not demonstrated this effect.[34] Lastly, none of the medications that our patient was taking are potent inhibitors of any of the cytochrome P450 enzymes.[35]
Summary. Although the etiology of TGA is not fully understood, we have indicated that TGA and TIA are separate entities. Nonetheless, a review of the literature has demonstrated cases where individuals experience amnesia without any neurological deficits—neither focal nor diffuse.[44–48] These examples exemplify the theory of the vertebrobasilar system’s role in TGA. They also show that TGA can, in fact, occur without any neurological deficits, let alone deficits specific to areas of the brain supplied by the vertebrobasilar system.
Thus, we posit that our patient’s symptoms were likely due to TGA (from basilar artery stenosis), which by definition, resolved within one day. Her treatment plan involved monitoring the resolution of her memory loss/psychoses. Though the likelihood of future recurrence is about five percent, no further treatment was warranted.[3]
Transient global amnesia and the neurobiology of delusions. Our patient’s emotional response during her TGA episode was consistent not only with “fear” of being in an “unfamiliar” situation, but inappropriate inferences about the “intent” of the medical staff (i.e., paranoid delusion).
The latter subsequently abated upon dissolution of her retrograde and anterograde amnesia (i.e., the secession of her TGA). Thus, we use this case to review and supplement the literature about the cognitive and neurobiological aspects of (paranoid) delusion formation.
While a complete review of fear learning is beyond the scope of this article, a basic understanding of this topic is necessary in navigating the bridge from TGA to delusion formation.
Neural circuitry in anxiety and paranoia. Affective learning involves a circuit incorporating amygdala, hippocampus, striatum, and prefrontal cortex (Figure 1). Dysfunctions within these nodes could engender fear in the wrong context, leading to maladaptive learning about the danger of adverse consequences. More particularly, the hippocampus is theorized to be essential for declarative memory (i.e., the conscious recall of specific experiences from one’s past [episodic memory][49] and context-dependent learning). Here, learned information becomes associated with context such that context can cue the relevant memories and behaviors.[50] The amygdala is crucial for fear learning. Toward that end, amygdala lesions in humans result in emotional blunting and reduced fear conditioning and abnormal activation of the amygdala in patients with temporal lobe epilepsy leads to a behavioral pattern reminiscent of fear.[51]

On a cognitive level, both anxiety and psychosis could arise from an aberrant assignment of novelty and salience to objects and associations. In anxiety states, the amygdala seems to have a specific role in the modulation of vigilance and attention to emotionally salient information,[51] and its hyperactivity has been proposed as a marker of negative affective bias in anxiety.[52]
Neuroimaging studies using tasks that involve salience processing indicate that the salience network involves midbrain dopamine neurons, their projections to the striatum, and the frontotemporal and frontoparietal cortical regions. There is converging evidence indicating that dopaminergic neurotransmission is essential for signaling reward and salience.[53] It has been proposed that during delusion formation, the normal process of context-driven novelty and salience attribution is usurped by an endogenously driven assignment of novelty and salience to stimuli.[54]
Thus, it has been proposed that paranoid thoughts may be arranged according to a “hierarchy” in which mild paranoia (e.g., fear of being talked about) is prevalent in the general population, whereas more severe paranoid thoughts (e.g., conspiracies) are most prevalent in clinical samples. The phenotypic expression on the fear-psychoses/paranoia continuum may be determined by the relative increase in mesolimbic dopaminergic neurotransmission. Anxiety/fear may be expressed due to mild-moderate increases in dopamine signaling, while in vulnerable individuals, moderate-severe accentuation in the same signaling pathway may culminate in psychoses/paranoid delusions.[55]
Neural signaling in anxiety and paranoia. The hippocampus contains local glutamatergic and gamma aminobutryic acid (GABA) (i.e., is activated by or secreting GABA) circuits. In the presence of negligible threat, hippocampus inhibits amygdala overactivity through inhibitory GABAergic (or anti-glutamatergic) neurotransmission.[56] Thus, diminution in hippocampal function can result in amygdalar disinhibition, precipitating “fear.”
TGA-induction of the anxiety/paranoia continuum: synthesizing cognitive descriptors, neural circuitry and signaling. TGA can be viewed as an illness of transient hippocampal insufficiency. Our patient’s transcranial PVL, consistent with basilar artery stenosis, may be pathogenically related to the development of her transient memory loss, or TGA. The hippocampus receives its vascular supply primarily from the posterior cerebral artery (PCA), which originates from the basilar artery (found to be stenotic in this patient), and to a lesser degree from the anterior choroidal artery (AChA), which originates from the internal carotid artery (also found to be stenotic in this patient). The PCA directly and by its branches contributes much more to the blood supply of the hippocampal formation than the AchA.[57] Subsequently, stenosis in the basilar artery and to a lesser degree the internal carotid artery may impact blood flow to the hippocampus.
In the case of our patient, during her TGA episode, she had no memory of events that both resulted in hospitalization or why she continued to require hospitalization, despite being informed on multiple occasions (i.e., retrograde amnesia, context-dependent-learning, and anterograde amnesia, respectively). This seemed to result in overgeneralization of fear/ “mistrust” of all hospital staff and the feeling of imminent “danger.” It seems clear that our patient’s “paranoia” could be accounted for parsimoniously, by appealing to an inappropriate engagement of the brain’s fear system.[54]
Please see Figure 2 for the pathobiology of how TGA could lead to the genesis of paranoid delusions.
Conclusion
In closing, our patient’s episode of TGA combined with her emotional and perceptual response lends credence to the proposal of a “fear/paranoia” circuit in the genesis of paranoid delusions—a circuit incorporating amygdala, frontal, and parietal cortices. Here, neutral or irrelevant stimuli, thoughts, and percepts come to engender fear and anxiety, while dysfunction in frontoparietal circuitry engenders inappropriate social predictions and maladaptive inferences about the intentions of others.[54]
Hippocampus relays information about contextual information based on past experiences and the current situation to amygdala, which the latter requires for threat assessment. Simply stated, in the absence of true threat, hippocampus, as well as ventromedial prefrontal (vMPFC) and anterior cingulate cortex (ACC), “inhibits” potential hyper-responsivity of amygdala.The hippocampus contains local glutamatergic and gamma aminobutryic acid (GABA). In the presence of negligible threat, hippocampus inhibits amygdala overactivity through inhibitory GABAergic (or anti-glutamatergic) neurotransmission.[56] Thus, diminutions in hippocampal function can result in amygdalar disinhibition, precipitating “fear.”
Additionally, there are extensive reciprocal connections between amygdala and the vMPFC and ACC. Glutamatergic efferents from both vMPFC and ACC likely synapse on ?-aminobutyric acid neurons and thus provide an important inhibitory input to the amygdala.[67]
Social cognition, which in part is responsible for the ability to represent and attribute affective and cognitive mental states to self and others, is localized to multiple structures within the brain. These regions include, but are not limited to, vMPFC/ACC, orbitofrontal cortex, temporo-parietal junction, superior temporal sulcus, amygdala, and striatum.[68]
Paranoid delusions/ideations are associated with excessive fear or anxiety. In regard to our patient’s phenomenology, paranoia may occur when uncertainty results from lack of contextual information (patient being unaware of why she was admitted to or required continued hospitalization) secondary to hypofunctioning of hippocampus during TGA. Decreased efferent GABAergic transmission disinhibits amygdala (development of fear, hypervigilance, or “paranoia.”) with downstream increase in midbrain dopaminergic transmission.
As paranoid thoughts are commonly about others,[54,69] inferring what people are thinking and feeling (theory of mind [TOM]) is an important aspect of human social cognition,[69] requiring access of episodic memories of past emotional experiences through intact hippocampal function. An inefficiency in the latter, such as during TGA, could explain the role of TOM impairment in paranoid delusions.[70] Our patient was extremely suspicious of the nursing/hospital staff and essentially was not able to infer the intentions/emotions of the staff, thus refusing laboratory/nursing care.
Dopamine has been intimated as the most popular candidate neurotransmitter associated with TOM.[71] It has been proposed that a disruption to the dopamine system itself could lead to the generation of erroneous predictions about the content of the mind of others, as is the case for patients with schizophrenia.[72]
References
1. Hodges JR, Warlow CP. Syndromes of transient amnesia: towards a classification. a study of 153 cases. J Neurol Neurosurg Psychiatry. 1990;53(10):834–843.
2. Bartsch T, Butler C. Transient amnesic syndromes. Nat Rev Neurol. 2013;9(2):86–97. Epub 2013 Jan 8.
3. Quinette P, Guillery-Girard B, Dayan J, et al. What does transient global amnesia really mean? review of the literature and thorough study of 142 cases. Brain. 2006;129(Pt 7): 1640–1658. Epub 2006 May 2.
4. Ott BR, Saver JL. Unilateral amnesic stroke. Six new cases and a review of the literature. Stroke. 1993;24(7):1033.
5. Yang Y1, Kim S, Kim JH. Ischemic evidence of transient global amnesia: location of the lesion in the hippocampus. J Clin Neurol. 2008;4(2):59–66.
6. Melo TP, Ferro JM, Ferro H. Transient global amnesia. a case control study. Brain. 1992;115 Pt 1:261–270.
7. Dalla Volta G, Zavarise P, Ngonga G, et al. Transient global amnesia as a presenting aura. Headache. 2014;54(3):551–552. Epub 2014 Feb 11.
8. Zorzon M, Antonutti L, Masè G, et al. Transient global amnesia and transient ischemic attack: natural history, vascular risk factors, and associated conditions. Stroke. 1995;26(9):1536–1542.
9. Lewis SL. Aetiology of transient global amnesia. Lancet. 1998;352(9125):397–399.
10. Inzitari D, Pantoni L, Lamassa M, et al. Emotional arousal and phobia in transient global amnesia. Arch Neurol. 1997;54(7):866–873.
11. Inouye SK, van Dyck CH, Alessi CA, et al. Clarifying confusion: the confusion assessment method. a new method for detection of delirium. Ann Intern Med. 1990;113(12):941–948.
12. Walsh RD, Wharen RE Jr, Tatum WO 4th. Complex transient epileptic amnesia. Epilepsy Behav. 2011;20(2):410–413.
13. Zeman AZ, Boniface SJ, Hodges JR. Transient epileptic amnesia: a description of the clinical and neuropsychological features in 10 cases and a review of the literature. J Neurol Neurosurg Psychiatry. 1998;64:435–443.
14. Butler CR, Bhaduri A, Acosta-Cabronero J, et al. Transient epileptic amnesia: regional brain atrophy and its relationship to memory deficits. Brain. 2009;132(Pt 2):357.
15. Khare S. Risk factors of transient ischemic attack: An overview. J Midlife Health. 2016;7(1):2–7.
16. Bilo L, Meo R, Ruosi P, et al. Transient epileptic amnesia: an emerging late-onset epileptic syndrome. Epilepsia. 2009;50 Suppl 5:58.
17. Milton F, Muhlert N, Pindus DM, et al. Remote memory deficits in transient epileptic amnesia. Brain. 2010;133(Pt 5):1368.
18. Gallassi R. Epileptic amnesic syndrome: an update and further considerations. Epilepsia. 2006;47 Suppl 2:103.
19. Caplan LR. Transient ischemic attack: definition and natural history. Curr Atheroscler Rep. 2006;8(4):276.
20. Förster A, Griebe M, Gass A, et al. Diffusion-weighted imaging for the differential diagnosis of disorders affecting the hippocampus. Cerebrovasc Dis. 2012;33(2):104–115. Epub 2011 Dec 14.
21. Sedlaczek O, Hirsch JG, Grips E, et al. Detection of delayed focal MR changes in the lateral hippocampus in transient global amnesia. Neurology. 2004;62:2165–2170.
22. Kidwell CS, Alger JR, Di Salle F, et al. Diffusion MRI in patients with transient ischemic attacks. Stroke. 1999;30(6):1174–1180.
23. Di Paola M, Moscatelli A, Bigler ED, et al. White matter changes in patients with hypoxic amnesia. 2011;17(1):46–56. Epub 2010 Oct 27.
24. Kirchhoff BA, Lugar HM, Smith SE, et al. Hypoglycaemia-induced changes in regional brain volume and memory function. Diabet Med. 2013;30(4):e151–156.
25. Silveri MM, Cohen-Gilbert J, Crowley DJ, et al. Altered anterior cingulate neurochemistry in emerging adult binge drinkers with a history of alcohol-induced blackouts. Alcohol Clin Exp Res. 2014;38(4):969–979. Epub 2014 Feb 11.
26. Mansour G, Abuzaid A, Bellamkonda P. I do not even remember what I smoked! A case of marijuana-induced transient global amnesia. Am J Med. 2014;127(11):e5–6. Epub 2014 Jul 11.
27. Welch KA. Neurological complications of alcohol and misuse of drugs. Pract Neurol. 2011;11(4):206–219.
28. Kimura S, Kumano T, Miyao S, et al. Herpes simplex encephalitis with transient global amnesia as an early sign. Intern Med. 1995;34(2):131–133.
29. Ely E, Inouye SK, Bernard GR, et al. Delirium in mechanically ventilated patients: validity and reliability of the Confusion Assessment Method for the Intensive Care Unit (CAM-ICU). JAMA. 2001;286(21):2703–2710.
30. Victor M, Adams RA, Collins GH. Wernick-Korsakoff syndrome related disorders due to alcoholism and malnutrition. Philadelphia: F A Davis; 1989.
31. Sutherland JJ, Daly TM, Liu X, Goldstein K, Johnston JA, Ryan TP. Co-prescription trends in a large cohort of subjects predict substantial drug-drug interactions. PLoS One. 2015;10(3):e0118991. doi: 10.1371/journal.pone.0118991. eCollection 2015.
32. Vermeeren A, Coenen AM. Effects of the use of hypnotics on cognition. Prog Brain Res. 2011;190:89–103. doi: 10.1016/B978-0-444-53817-8.00005-0.
33. Jamolowicz AI, Chen HY, Panegyres PK. Statins and memory loss: an Australian experience. Australas Med J. 2015;8(3):73-9. doi: 10.4066/AMJ.2015.2014. eCollection 2015.
34. Cooke JD, Cavender HM, Lima HK, Grover LM. Antidepressants that inhibit both serotonin and norepinephrine reuptake impair long-term potentiation in hippocampus. Psychopharmacology (Berl). 2014;231(23):4429–4441. doi: 10.1007/s00213-014-3587-1. Epub 2014 Apr 30.
35. Kerr KP, Mate KE, Magin PJ, et al. The prevalence of co-prescription of clinically relevant CYP enzyme inhibitor and substrate drugs in community-dwelling elderly Australians. J Clin Pharm Ther. 2014;39(4):383–389. doi: 10.1111/jcpt.12163. Epub 2014 Apr 7.
36. Brandt J, Van Gorp WG. Functional (“psychogenic”) amnesia. Semin Neurol. 2006;26(3):331–340.
37. Santos S, López del Val J, Tejero C, et al. Transient global amnesia: a review of 58 cases. Rev Neurol. 2000;30(12):1113–1117.
38. Lewis SL. Aetiology of transient global amnesia. Lancet. 1998;352(9125):397–399.
39. Pantoni L, Lamassa M, Inzitari D. Transient global amnesia: a review emphasizing pathogenic aspects. Acta Neurol Scand. 2000;102:275–283.
40. Schreiber SJ, Doepp F, Klingebiel R, et al. Internal jugular vein valve incompetence and intracranial venous anatomy in transient global amnesia. J Neurol Neurosurg Psychiatry. 2005;76:509–513.
41. Sander D, Winbeck K, Etgen T, et al. Disturbance of venous flow patterns in patients with transient global amnesia. Lancet. 2000;356:1982–1984.
42. Maalikjy Akkawi N, Agosti C, Anzola GP, et al. Transient global amnesia: a clinical and sonographic study. Eur Neurol. 2003;49:67–71.
43. Döhring J, Schmuck A, Bartsch T. Sturctural and functional correlate of TGA. Stress-related factors in the emergence of transient global amnesia with hippocampal lesions. Front Behav Neurosci. 2014;8:287.
44. Jensen TS, De Fine Olivarius B. Transient global amnesia as a manifestation of transient cerebral ischemia. Acta Neurol Scand. 1980;61(2):115–124.
45. Otsuka Y, Tsuchikawa S, Mitake A, et al. [A case of transient global amnesia (TGA) showing bilateral hippocampal hypoperfusion by a new SPECT analyzing system, eZIS]. Rinsho Shinkeigaku. 2002;42(10):977–979.
46. Taylor RA, Wu GF, Hurst RW, et al. Transient global amnesia heralding basilar artery thrombosis. Clin Neurol Neurosurg. 2005;108(1):60–62. Epub 2004 Dec 21.
47. Schmidtke K, Reinhardt M, Krause T. Cerebral perfusion during transient global amnesia: findings with HMPAO SPECT. J Nucl Med. 1998;39(1):155–159.
48. Takeuchi R, Matsuda H, Yoshioka K, et al. Cerebral blood flow SPET in transient global amnesia with automated ROI analysis by 3DSRT. Eur J Nucl Med Mol Imaging. 2004;31(4):578–589. Epub 2004 Jan 14.
49. Knierim JJ. Neural representations of location outside the hippocampus. Learn Mem. 2006;13(4):405–415.
50. Smith DM, Mizumori SJ. Learning-related development of context-specific neuronal responses to places and events: the hippocampal role in context processing. J Neurosci. 2006;26(12):3154–3163.
51. Phillips ML, Drevets WC, Rauch SL et al. Neurobiology of emotion perception I: The neural basis of normal emotion perception. Biol Psychiatry. 2003;54(5):504–514.
52. Robinson OJ, Overstreet C, Allen PS, et al. The role of serotonin in the neurocircuitry of negative affective bias: serotonergic modulation of the dorsal medial prefrontal-amygdala ‘aversive amplification’ circuit. Neuroimage. 2013;78:217–223.
53. Mishara AL, Fusar-Poli P. The phenomenology and neurobiology of delusion formation during psychosis onset: Jaspers, Truman symptoms, and aberrant salience. Schizophr Bull. 2013;39(2):278–286.
54. Phillips ML, Drevets WC, Rauch SL et al. Neurobiology of emotion perception II: Implications for major psychiatric disorders. Biol Psychiatry. 2003;54(5):515–528.
55. Savulich G, Freeman D, Shergill S, et al. Interpretation biases in paranoia. Behav Ther. 2015;46(1):110–124.
56. Salloway SP, Blitz A. Introduction to functional neural circuitry. In: Kaplan GB, Hammer RP Jr, eds. Brain Circuitry and Signaling in Psychiatry: Basic Science and Clinical Implications. Washington, DC: American Psychiatric Publishing, Inc; 2002:1–30.
57. Erdem A, Yaargil G, Roth P. Microsurgical anatomy of the hippocampal arteries. J Neurosurg. 1993;79(2):256–265.
58. Corlett PR, Taylor JR, Wang XJ, et al. Toward a neurobiology of delusions. Prog Neurobiol. 2010;92(3):345–369.
59. Armstrong RC, Mierzwa AJ, Marion CM, Sullivan GM. White matter involvement after TBI: Clues to axon and myelin repair capacity. Exp Neurol. 2015 Feb 16. pii: S0014-4886(15)00034-5. doi: 10.1016/j.expneurol.2015.02.011. [Epub ahead of print]
60. Nuwer MR, Hovda DA, Schrader LM, Vespa PM. Routine and quantitative EEG in mild traumatic brain injury. Clin Neurophysiol. 2005;116(9):2001–2025.
61. Haegerich TM, Dahlberg LL, Simon TR, et al. Prevention of injury and violence in the USA. Lancet. 2014;384(9937):64–74. doi: 10.1016/S0140-6736(14)60074-X. Epub 2014 Jul 1.
62. Regier DA, Farmer ME, Rae DS, et al. Comorbidity of mental disorders with alcohol and other drug abuse. Results from the Epidemiologic Catchment Area (ECA) Study. JAMA. 1990;264(19):2511–2518.
63. Vroegop MP, Franssen EJ, van der Voort PH, et al. The emergency care of cocaine intoxications. Neth J Med. 2009;67(4):122–126.
64. Daras M, Tuchman AJ, Marks S. Central nervous system infarction related to cocaine abuse. Stroke. 1991;22(10):1320–1325.
65. Reid MS, Flammino F, Howard B, et al. Topographic imaging of quantitative EEG in response to smoked cocaine self-administration in humans. Neuropsychopharmacology. 2006;31(4):872–884.
66. Bolla KI, Eldreth DA, Matochik JA, Cadet JL. Neural substrates of faulty decision-making in abstinent marijuana users. Neuroimage. 2005;26(2):480–492. Epub 2005 Mar 23.
67. Davidson RJ. Anxiety and affective style: role of prefrontal cortex and amygdala. Biol Psychiatry. 2002;51(1):68–80.
68. Abu-Akel A, Shamay-Tsoory S. Neuroanatomical and neurochemical bases of theory of mind. Neuropsychologia. 2011;49(11):2971–2984.
69. Saxe R. Uniquely human social cognition. Curr Opin Neurobiol. 2006;16(2):235–239.
70. Calarge C, Andreasen NC, O’Leary DS. Visualizing how one brain understands another: a PET study of theory of mind. Am J Psychiatry. 2003;160(11):1954–1964.
71. Xia H, Wu N, Su Y. Investigating the genetic basis of theory of mind (ToM): the role of catechol-O-methyltransferase (COMT) gene polymorphisms. PLoS One. 2012;7(11):e49768.
72. Abu-Akel A. The neurochemical hypothesis of ‘theory of mind.’ Med Hypotheses. 2003;60(3):382–386.