
by David R. Spiegel, MD, and Kathryn Webb, MD
Drs. Spiegel and Webb are from Eastern Virginia Medical School, Department of Psychiatry and Behavioral Sciences in Norfolk, Virginia.
Innov Clin Neurosci. 2012;9(11–12):31–38
Funding: There was no funding for the development and writing of this article.
Financial Disclosures: The authors do not have any conflicts of interest relevant to the content of this article.
Key words: Hyperemesis gravidarum, neurogastroenterology, gabapentin
Abstract: Hyperemesis gravidarum occurs in 0.3 to 10 percent of pregnant women, with a 0.8 percent hospital admission rate. While older theories supported the psychosocial model as a cause for hyperemesis gravidarum, more recent studies have shown significant data to support a biological etiology. Hyperemesis gravidarum has serious complications including include increased risk for miscarriage, low birth weight infants, dehydration, Wernicke’s encephalopathy, secondary depression, and negative attitudes toward a consecutive pregnancy. Because of these life-threatening complications and complexity of the disease, it is important to treat both somatic and psychosocial causes of hyperemesis gravidarum to provide the best care for the patient. This paper presents a case of a woman with anxiety symptoms who was experiencing severe nausea and vomiting since Week 2 of pregnancy, with minimal reduction of these symptoms on standard medications utilized in hyperemesis gravidarum. The patient had marked reduction of nausea and vomiting with adjunctive gabapentin. After a brief review of relevant neurogastroenterology, we discuss a possible mechanism for the added gabapentin.
INTRODUCTION
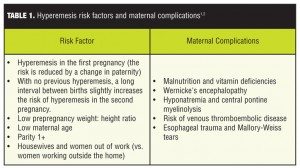
Hyperemesis gravidarum (HG) is defined as extreme vomiting during pregnancy associated with electrolyte imbalance, five-percent weight loss, or ketosis. It is estimated that this condition occurs in 0.3 to 10 percent of pregnant women, with a 0.8 percent hospital admission rate. The average onset of HG is within four weeks of gestation; it peaks at nine weeks gestational age and usually resolves by week 20 of pregnancy. Risk factors (Table 1) associated with HG are primarily medical and fetal factors that are not easily modifiable, but their identification may be useful in determining those women at high risk for developing HG.[1] Maternal complications Table 1) include malnutrition and vitamin deficiencies and central pontine myelinolysis.[2] Treatment for HG (see Table 2 for more complete listing and dosing) includes antinausea/vomiting medications (including, 5-hydroxytryptamine3-receptor antagonists) and rehydration in extreme cases.[3] This paper presents a case of a woman who experienced symptoms of HG since Week 2 of pregnancy and was refractory to conventional antiemetics normally utilized in HG. Her symptoms were attenuated by addition of gabapentin (GPN).
Case Report
Our patient was a 34-year-old woman, gravdia 4, parity 1. Her first two pregnancies ended with a spontaneous abortion at three months; her next pregnancy ended at 32 weeks, and resulted in a live male child. Her current pregnancy was at 23 weeks gestational age. During this pregnancy, the patient experienced extreme nausea and vomiting (N/V) since Week 2 of pregnancy and was officially diagnosed with HG at Week 6 of pregnancy. She was taking both ondansetron and promethezine since diagnosed with HG, and ginger was added two weeks later, with minimal effectiveness. Her Motherisk Pregnancy-unique Quantification of Emesis and Nausea (PUQE) scale was 15 on admission (PUQE scale ranges from 3 [no symptoms] to 15 [maximal symptoms]).[4] Her current hospitalization was due to placenta previa (PPr) causing vaginal bleeding. A complete blood count showed a slightly increased white blood cell count and decreased hemoglobin and hematocrit, likely due to bleeding from PPr. Save decreased total protein and albumin, a complete metabolic panel was within normal limits. We were consulted to address the patient’s anxiety.
It was found that the patient had many current stressors, including taking care of her 11-year–old autistic son. Her Hamiliton Anxiety Scale (HAM-A)5 score equaled 22 on admission. The patient denied any current or history of depressed or manic episodes or psychotic symptoms. She did have a history of cocaine use which stopped 15 years ago. She endorsed using alcohol; however, the patient had not been drinking during the past one year. Her urine drug screen and blood alcohol were negative on admission.
We began our patient was on gabapentin 100mg TID, increased to 300mg TID after two days. After three days of the increased gabapentin dosage, her Motherisk PUQE and HAM-A scores were 8 and 6, respectively.[4,5] She was discharged from the hospital and instructed to continue the gabapentin 300mg TID. The patient subsequently delivered two weeks later, at gestational age of 27 weeks, an Apgar score 6 of 8 at five minutes and was two pounds, seven ounces. Our patient’s readmission Motherisk PUQE scale score was 5. Additionally, while the obstetrician thought this premature delivery was due obstetrical complication of PPr, gabapentin administration could not be ruled out.
Discussion
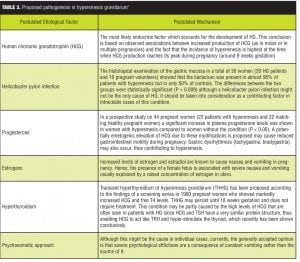
The cause or causes of HG remain unclear but may be attributed to hormones, gastrointestinal (GI) dysfunction, thyrotoxicosis, serotonin, hepatic abnormalities, autonomic nervous dysfunction, nutritional deficiencies, asthma, allergies, Helicobacter pylori infection, or psychosomatic causes (Table 3).[7] Few large trials have identified the optimal therapy for this condition; however, to the best of the authors’ knowledge, only one other case of HG has been (successfully) treated and reported with GPN.[8]
First, GPN has received a pregnancy category C from the Food and Drug Administration (FDA). Second, reports on the consequences of prenatal GPN exposure are limited and inconclusive and need be considered if prescribing this medication during pregnancy. In one study comprising 51 infants,[41] no increased risk for fetal malformations was found. However, it should be noted that a study in six women suggested an active transplacental transport of GPN with accumulation in the fetus.[9]
One hypothesis concerning the pathogenesis of HG involves an interaction of estrogen/progesterone on neural networks innervating the gastrointestinal tract. Thus, while extensively reviewed elsewhere,[10] a brief review of neurogastro-enterology is warranted. The gastrointestinal (GI) tract differs from all other peripheral organs in that it has an extensive intrinsic nervous system, termed the enteric nervous system (ENS), that can control functions of the intestine, even if the latter is completely separated from the central nervous system (CNS). The ENS, however, is not autonomous.
The brain–gut axis roughly consists of three parts: the enteric nervous system, the autonomic nervous system (ANS), and the CNS. The ANS transfers information from the gut to the brain via vagal and spinal afferent pathways. At brain level, the information is processed (affective and cognitive dimensions are added to it). Finally, the brain sends information back to the gut via the ANS (parasympathetic and sympathetic efferents).[11]
More specifically, visceral sensory information from the stomach is transmitted through the afferent vagus into the brainstem via a glutamatergic synapse at the level of the nucleus of the tractus solitarius (NTS). NTS neurons project to, among other areas, the adjacent dorsal motor nucleus of the vagus (DMV) using mainly gamma-aminobutyric acid (GABA), glutamate, and catecholamines as neurotransmitters. Interestingly, there appears to be a spatial organization of vagal preganglionic (parasympathetic) neurons in the DMV. The rostral DMV preferentially innervates a cholinergic excitatory pathway responsible for gastric contraction while caudal DMV neurons are efferent to an inhibitory pathway responsible for gastric relaxation. Dorsal motor nucleus of the vagus neurons are, a priori, cholinergic and innervate postganglionic neurons located within the stomach. In the stomach, postganglionic parasympathetic neurons form two distinct pathways: 1) an excitatory cholinergic pathway that increases gastric tone, motility and secretion via activation of muscarinic cholinergic receptors and 2) an inhibitory nonadrenergic, non-cholinergic (NANC) pathway that inhibits gastric functions via release of predominantly nitric oxide (NO) or vasoactive intestinal polypeptide (VIP). Ultimately, gastric tone represents a balance between the excitatory cholinergic and the inhibitory NANC pathways. For example, gastric functions may be inhibited, i.e., decreased motility/delayed gastric emptying, either by activation of the NANC pathway or by inhibition of the tonic cholinergic pathway (Figure 1).[12]
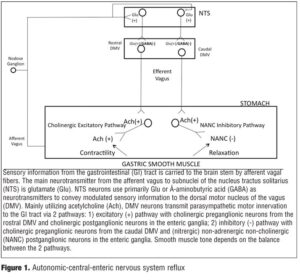
Thus, within the GI system, neurotransmitters, neuromodulators, and neurohumoral agents appear capable of modulating vago-vagal reflexes at multiple sites, from altering the phenotype of vagal afferent neurons to regulating the release of glutamate from the central terminals of vagal afferents to regulating the strength of GABAergic inputs onto gastric projecting vagal efferent neurons. Interestingly, despite the presence of ENS in the stomach, the CNS provides the primary control of propulsion; indeed, if the vagi are severed, the stomach is paralyzed and food is no longer propelled.[12]
During pregnancy, some[13] but not all,[14] studies report decreased motility of the gastrointestinal tract leading to a delay in gastric emptying and an increase in colonic transit time. Whether the rise in estradiol or progesterone is responsible for this effect is controversial; however, release of nitric oxide (NO) by neuronal NO synthase (nNOS) found in NANC neurons has been shown to be an important factor controlling/ decreasing gastrointestinal motility and transit time. Studies suggest that the increase in sex steroids during pregnancy increases nitergic activity supporting a possible pregnancy-related explanation for gastroparesis.[13] It is unknown whether sex steroids affect GABA or glutamatergic functioning upstream in the GI vago-vagal reflex/pathway.
An alternative theory to explain the pathophysiology of HG stems from the gastrointestinal musculature as a self-excitable electrical syncytium consisting of interstitial cells of Cajal (ICCs) that function as pacemakers for the gastric musculature and the intestinal circular muscle coat. The ICCs are a nonneural pacemaker system of electrical slow waves (rhythmic depolarizations) that occur at a frequency of three cycles/minute(cpm).[15]
Similar to the heart, ectopic pacemakers in other parts of the stomach (primarily the antrum) may generate regular or disorganized rhythms in a number of clinical conditions. Bradygastria develops when the normal dominant pacemaker fails and other oscillatory sites in the gastric body generate rhythmic depolarizations at frequencies less than 2cpm. With bradygastria, the contractile efficiency of the stomach is reduced due to a decrease in the number of antral contractions during fasting and the postprandial period. Tachygastria develops when a rival pacemaker, usually in the antrum, generates an oscillatory pattern at an abnormally high frequency (>4cpm) that overrides the rest of the stomach. Although retrograde depolarization propagates at a high frequency with tachygastria, retrograde motor activity rarely develops because the electrical activity is of insufficient amplitude to induce contraction. Hence, during tachygastria, the stomach is atonic. Not infrequently, the ectopic pacemaker activity is highly unstable both in frequency and in location, which results in the development of tachybradyarrhythmia.[16]
A number of clinical conditions have been found to be associated with gastric slow-wave rhythm disturbances in the antral pump that may relate to the induction of N/V, including HG.[17] Increases in b-HCG during pregnancy leads to increased levels of estrogen and progesterone. Both the former and the latter have been reported to result not only in N/V but also to be mediators of gastric slow-wave rhythm disruption.[18] Conversely, pregnant women without active N/V do not exhibit slow-wave dysrhythmias.[16]
Our patient’s symptoms of N/V significantly improved during her hospitalization course with gabapentin. Gabapentin was first introduced as an antiepileptic drug but has more recently been used in the treatment of a variety of pain states. Although structurally related GABA, a major inhibitory neurotransmitter in the CNS, gabapentin is functionally inactive at GABA-A, GABA-B, or benzodiazepine receptors. The predominant mechanism of action of gabapentin which explains its pharmacological profile is the inhibition of calcium currents via high-voltage-activated channels containing the alpha2delta subunit, leading in turn to reduced neurotransmitter release and attenuation of postsynaptic excitability. Emerging data supports a role for the alpha2delta subunit in neurotransmission in the enteric nervous system, but the functional importance of these observations has yet to be fully elucidated.[19]
There are many possible explanations that could have resulted in our patient’s attenuation of HG symptoms to gabapentin. By suggesting that the symptoms for HG could, in part, be accounted for by delayed gastric emptying (or gastroparesis), we briefly will review the latter.
The hallmark of gastroparesis is delayed gastric emptying in the absence of a mechanical obstruction. Gastroparesis occurs as a result of failure of the normal gastric emptying process, which, in health, is controlled through a coordinated interaction between the ANS, especially vagal nerve neuropathy, the ENS, and the “pacemaker cells,” i.e., ICC, which are essential for the propagation of gastric rhythm. When these interactions fail and gastroparesis results, the latter may be characterized by one or more abnormalities in gastric motility, including fundal and/or antral hypomotility and gastric arrhythmias.[20] Remember that both fundal and antral contractility are dependent on the cholinergic excitatory pathway of the vago-vagal reflex (VVR) and that ultimate gastric motility depends on a balance between the excitatory and inhibitory pathways of the VVR.[12]
We thus posit that in gastroparesis, the VVR is skewed toward gastroinhibition. By attenuating glutamate release from afferent vagus transmission to NTS, DMV neurons become disinhibited vis-à-vis decreasing GABA transmission from NTS to DMV. As such efferent vagus transmission is modified such that either gastroexcitatory pathways become augmented and/or gastroinhibitory pathways are attenuated, resulting in “pro-kinetic” activity of stomach musculature. This could increase contractility at the fundus and/or antrum, potentially resulting in improved gastric emptying, and a lessening of profuse N/V that can occur in HG.
Alternatively, normalization of gastric dysrhythmias (GD) has proven effective in treating N/V associated with GD. Both central and local inhibition of cholinergic activity has been proposed as mediators of this effect;[21] however, intracellular calcium (as well as various receptors) handling has a critical role in the generation of pacemaker activity in the gut.[22] Nonetheless, gabapentin has been demonstrated to inhibit release of acetylcholine (at least, in the CNS),[23] possibly resulting in a return of the dominant slow wave rhythm of the ICC.
Lastly, while an association between HG and psychiatric illness may be present, data do not support a unidirectional link of psychological distress causing HG. It remains unclear whether psychiatric illness leads to production, whether the stress of HG symptoms leads to psychiatric impairment, or whether these conditions are separate but interact. Furthermore, and while limited by very small sample sizes, a prospective case control study found that while suffering from HG, women showed more anxiety symptoms. Interestingly, during the postpartum period, there were no significant differences between groups.[24]
Nonetheless, in one recent study, the current prevalence rate of any mood or anxiety disorder in pregnant women with and without HG was 46.2 percent and 14.4 percent, respectively. Large-scale epidemiological studies have indicated that 4.7 to 18.3 percent of women in the general population have at least one current Axis I psychiatric disorder. In this same study, the prevalence rate of any mood or anxiety disorder prior to the pregnancy in women with HG was higher than that in the control subjects; however, 29.5 percent of HG women with psychiatric diagnosis described an onset of psychiatric disorder after the onset of HG. These authors conclude that mood and anxiety disorders may be associated with the occurrence of HG in pregnant women. Finally, and as found in our patient, this study demonstrated that overall prevalence rate of anxiety disorders, but not mood disorders, was significantly higher in the women with HG than in the control pregnant women. Interestingly, while these authors do not ascribe causality of mood or anxiety disorders to HG, they state that “further studies also should examine whether successful treatment of psychiatric disorders accompanying HG may contribute to a resolution of HG symptoms, particularly in the cases resistant to medical treatment.”[25]
Thus, while GPN could be effective in treating HG vis-à-vis modifying underlying ANS pathology and/or GD, there are reports of successful treatment of anxiety with gabapentin.[18] Therefore, the role of alleviating anxiety in our patient, (with anxiety in and of itself associated with nausea and vomiting during pregnancy[26]) could result in improvement in nausea and vomiting. Furthermore, in patients with functional dyspepsia presenting with vomiting, those patients who scored for anxiety showed significantly greater antral meal retention than patients without anxiety.[27] Hence, successful treatment of anxiety with GPN could directly improve gastric emptying.
Among the medical concerns of HG, symptomatic treatment of nausea and vomiting, correction of dehydration and electrolyte imbalance, and prevention of complications of the disease remains the mainstay. Dehydrated and ketotic women require admission. Therapy with intravenous fluids for correction of dehydration is the mainstay of management. The volume of fluid should replenish the deficit along with the loss through vomiting as well as meet normal fluid and electrolyte requirements. Fluid replacement is tailored to ketonuria or electrolytes and stopped once these are equalized and a normal diet is resumed.[1]
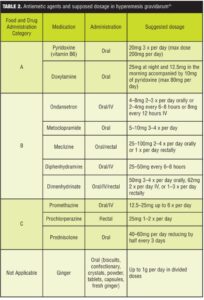
As for the “target symptoms” of nausea and vomiting, a recent Cochrane review found a lack of high-quality evidence to back up any advice on which interventions to use.[28] The American College of Obstetricians and Gynecologists (ACOG) has published an algorithm for the management of nausea and vomiting in pregnancy.[29] The ACOG currently recommends that a combination of oral pyridoxine hydrochloride (vitamin B6) and doxylamine succinate be used as first-line treatment for HG, if pyridoxine monotherapy does not relieve symptoms. Pyridoxine is a water soluble vitamin that is involved in the metabolism of amino acids, lipids, and carbohydrates. Doxylamine directly inhibits the action of histamine at the H1-receptor, acts indirectly at the vestibular system, and exhibits some inhibition of muscarinic receptors to decrease stimulation of the vomiting center. If the patient has a higher BMI, she may take up to 8 to 12 tablets a day without increasing maternal adverse effects, fetal risk, degree of tiredness, and birth defects. These higher doses appear to be more efficacious.[30]
After initiating treatment of HG with a combination of doxylamine and pyridoxine, breakthrough nausea and vomiting can be treated with the addition of a different antihistamine or a dopamine antagonist. Dimenhydrinate is an H1-receptor antagonist that is widely used for the treatment of NVP. In addition, it is often useful to initiate treatment with an H2-receptor antagonist as their safety and efficacy are evident when using these agents for treating the reflux and heartburn symptoms associated with NVP. Cimetidine, ranitidine, or famotidine are H2-receptor blockers that can be used especially if the patient has a history GERD, gastroduodenal ulcers, or other GI disease.[30]
Second-line treatment includes phenothiazines. The latter, such as prochlorperazine, are dopamine antagonists and inhibit vomiting by inhibiting the chemoreceptor trigger zone along with a direct action on the gastrointestinal tract D2 receptors. There have been case reports of cleft palate, skeletal, limb, and cardiac abnormalities with its use during pregnancy. The higher doses used for antipsychotic effect have been associated with temporary extrapyramidal effects postnatally, but the doses and duration used for antiemetic treatment are much lower.[1] Furthermore, the literature has numerous case reports of side effects of prochlorperazine, including tardive dyskinesia; however, increased duration and total cumulative dose of prochlorperazine increases the probability of tardive dykinesia.[31]
Additionally, the 5-HT3 receptor antagonist, ondansetron, also has a central chemoreceptor inhibition as well as a peripheral action on the small bowel and vagus nerve which inhibits vomiting.[1] Despite a lack of evidence on its use in pregnancy, data to date have been favorable. There has been no evidence of teratogenicity in animal studies even at doses significantly higher than that used in humans or in case reports of use in the first trimester.[30]
Metoclopramide, another second line-treatment for HG, stimulates gastric motility by affecting multiple receptor systems within the gut wall. Most importantly, it is an antagonist of the dopamine D2 receptor subtype. The anti-emetic effects of metoclopramide result from inhibition of D2 and 5-HT3 receptors within the chemoreceptor trigger zone. The risk of tardive dyskinesia with use of prolonged or high (cumulative) doses of metoclopramide prompted the United States Food and Drug Administration’s (FDA) black box warning regarding the use of metoclopramide and the risk of tardive dyskinesia. The warning continues that treatment for more than 12 weeks should be avoided;[3] however, one recent review showed that the risk of tardive dyskinesia from metoclopramide use is likely to be less than one percent, much less than the estimated 1- to 10-percent risk suggested in national guidelines.[32]
Finally, the use of corticosteroids for HG has received mixed reviews. Nonetheless, its antiemetic mechanism is not clear. It is suggested that corticosteroids may inhibit central prostaglandin synthesis, which is related to the triggering of emesis, or decrease serotonin turnover in the central nervous system. Alternatively, corticosteroids might exert an antiemetic effect through their action on the chemoreceptor trigger zone located in the brainstem. Numerous glucocorticoid receptors are found in the nucleus of the solitary tract, area postrema, and raphe nuclei. These nuclei are well known to have considerable neuronal activities in the regulation of nauseating and vomiting response. The preferred glucocorticoids during pregnancy are hydrocortisone or prednisone, because the placenta readily inactivates them.[33] Their use in pregnancy, however, has been associated with a small but significant increase in the incidence of oral cleft in a study published by Motherisk.[42] As steroid treatment for HG remains controversial, the drug should be reserved for women with prolonged or severe symptoms that are unresponsive to other treatments.[34]
Briefly, with respect to fetal outcomes, the reported effects of HG on birthweight and gestational age are conflicting, with some studies reporting shorter gestational ages and lower birthweights associated with HG pregnancies[35] and with others reporting no effects.[36] Nonetheless, a recent meta-analysis reported that women with HG are more likely to have a baby with low birthweight (LBW),
Conclusion
In closing, HG can potentially lead to significant morbidity in both mother and fetus. While there are studies demonstrating the efficacy of adjunctive diazepam in the treatment of HG,[37,38] this should be tempered against diazepam’s FDA pregnancy category D rating.[39] Thus, while certainly not first-line therapy, we feel that the treatment of HG with adjunctive GPN, especially in patients with comorbid anxiety symptoms, could be investigated further in the management of treatment refractory HG.
References
1. Sonkusare S. The clinical management of hyperemesis gravidarum. Arch Gynecol Obstet. 2011 Jun;283(6):1183–1192.
2. Ismail SK, Kenny L. Review on hyperemesis gravidarum. Best Pract Res Clin Gastroenterol. 2007;21(5):755–769.
3. Niebyl JR. Clinical practice. Nausea and vomiting in pregnancy. N Engl J Med. 2010;363(16):1544–1550.
4. Koren G, Boskovic R, Hard M, et al. Motherisk-PUQE (pregnancy-unique quantification of emesis and nausea) scoring system for nausea and vomiting of pregnancy. Am J Obstet Gynecol. 2002;186(5 Suppl Understanding):S228–S231.
5. Hamilton, M. The Assessment of Anxiety States by Rating. Br J Med Psychol. 1959;32:50–55.
6. Apgar V. A proposal for a new method of evaluation of the newborn infant. Curr Res Anesth Analg. 1953;32:260–267.
7. Wegrzyniak LJ, Repke JT, Ural SH. Treatment of hyperemesis gravidarum. Rev Obstet Gynecol. 2012;5(2):78–84.
8. Guttuso T Jr, Robinson LK, Amankwah KS. Gabapentin use in hyperemesis gravidarum: a pilot study. Early Hum Dev. 2010;86(1):65–66.
9. Reimers A, Brodtkorb E. Second-generation antiepileptic drugs and pregnancy: a guide for clinicians. Expert Rev Neurother. 2012;12(6):707–717.
10. Grundy D, Al-Chaer ED, Aziz Q, et al. Fundamentals of neurogastroenterology: basic science. Gastroenterology. 2006;130(5):1391–1411.
11. Van Oudenhove L, Demyttenaere K, Tack J, Aziz Q. Central nervous system involvement in functional gastrointestinal disorders. Best Pract Res Clin Gastroenterol. 2004;18(4):663–680.
12. Browning KN, Travagli RA. Plasticity of vagal brainstem circuits in the control of gastric function. Neurogastroenterol Motil. 2010;22(11):1154–1163.
13. Shah S, Nathan L, Singh R, et al. E2 and not P4 increases NO release from NANC nerves of the gastrointestinal tract: implications in pregnancy. Am J Physiol Regul Integr Comp Physiol. 2001;280(5):R1546–1554.
14. Maes BD, Spitz B, Ghoos YF, et al. Gastric emptying in hyperemesis gravidarum and non-dyspeptic pregnancy. Aliment Pharmacol Ther. 1999;13(2):237–243.
15. Wood JD. Neuropathophysiology of functional gastrointestinal disorders. World J Gastroenterol. 2007;13(9):1313–1332.
16. Owyang C, Hasler WL. Physiology and pathophysiology of the interstitial cells of Cajal: from bench to bedside. VI. Pathogenesis and therapeutic approaches to human gastric dysrhythmias. Am J Physiol Gastrointest Liver Physiol. 2002;283(1):G8–G15.
17. Grundy D, Al-Chaer ED, Aziz Q, et al. Fundamentals of neurogastroenterology: basic science. Gastroenterology. 2006;130(5):1391–1411.
18. Walsh JW, Hasler WL, Nugent CE, Owyang C. Progesterone and estrogen are potential mediators of gastric slow-wave dysrhythmias in nausea of pregnancy. Am J Physiol. 1996;270(3 Pt 1):G506–G514.
19. Gale JD, Houghton LA. Alpha 2 delta ligands, gabapentin and pregabalin: What is the evidence for potential use of these ligands in irritable bowel syndrome. Front Pharmacol. 2011;2:28.
20. Keld R, Kinsey L, Athwal V, Lal S. Pathogenesis, investigation and dietary and medical management of gastroparesis. J Hum Nutr Diet. 2011 Oct;24(5):421–430.
21. Kim TW, Koh SD, Ordög T, et al. Muscarinic regulation of pacemaker frequency in murine gastric interstitial cells of Cajal. J Physiol. 2003 Jan 15;546(Pt 2):415–425.
22. Takaki M. Gut pacemaker cells: the interstitial cells of Cajal (ICC). J Smooth Muscle Res. 2003;39(5):137–161.
23. Tzellos TG, Papazisis G, Toulis KA, et al. Alpha2delta ligands gabapentin and pregabalin: future implications in daily clinical practice. Hippokratia. 2010;14(2):71–75.
24. Kim DR, Connolly KR, Cristancho P, et al. Psychiatric consultation of patients with hyperemesis gravidarum. Arch Womens Ment Health. 2009;12(2):61–67.
25. Uguz F, Gezginc K, Kayhan F et al. Is hyperemesis gravidarum associated with mood, anxiety and personality disorders: a case-control study. Gen Hosp Psychiatry. 2012;34(4):398–402.
26. Jahangiri F, Hirshfeld-Cytron J, Goldman K, et al. Correlation between depression, anxiety, and nausea and vomiting during pregnancy in an in-vitro fertilization population: a pilot study. J Psychosom Obstet Gynaecol. 2011;32(3):113–118.
27. Lorena SL, Tinois E, Brunetto SQ, et al. Gastric emptying and intragastric distribution of a solid meal in functional dyspepsia: influence of gender and anxiety. J Clin Gastroenterol. 2004;38(3):230–236.
28. Matthews A, Dowswell T, Haas DM, et al. Interventions for nausea and vomiting in early pregnancy. Coch Data Syst Rev. 2010;(9):CD007575.
29. ACOG (American College of Obstetrics and Gynecology) practice bulletin: nausea and vomiting of pregnancy. Obstet Gynecol 2004;103:803–814.
30. Clark SM, Costantine MM, Hankins GD. Review of NVP and HG and early pharmacotherapeutic intervention. Obstet Gynecol Int. 2012;2012:252676. Epub 2011 Nov 24.
31. Olsen JC, Keng JA, Clark JA. Frequency of adverse reactions to prochlorperazine in the ED. Am J Emerg Med. 2000;18(5):609–611.
32. Rao AS, Camilleri M. Review article: metoclopramide and tardive dyskinesia. Aliment Pharmacol Ther. 2010;31(1):11–19.
33. Bondok RS, El Sharnouby NM, Eid HE, Abd Elmaksoud AM. Pulsed steroid therapy is an effective treatment for intractable hyperemesis gravidarum. Crit Care Med. 2006;34(11):2781–2783.
34. Ebrahimi N, Maltepe C, Einarson A. Optimal management of nausea and vomiting of pregnancy. Int J Womens Health. 2010;2:241–248.
35. Veenendaal M, van Abeelen A, Painter R, et al. Consequences of hyperemesis gravidarum for offspring: a systematic review and meta-analysis. BJOG 2011;118:1302–1313.
36. Kuru O, Sen S, Akbay O, et al. Outcomes of pregnancies complicated by hyperemesis gravidarum. Arch Gynecol Obstet. 2012;285(6):1517–1521.
37. Ditto A, Morgante G, la Marca A et al. Evaluation of treatment of hyperemesis gravidarum using parenteral fluid with or without diazepam. A randomized study. Gynecol Obstet Invest. 1999;48(4):232–236.
38. Tasci Y, Demir B, Dilbaz S, et al. Use of diazepam for hyperemesis gravidarum. J Matern Fetal Neonatal Med. 2009;22(4):353–356.
39. Drug Information Online. Diazepam. http://www.drugs.com/diazepam.html. Accessed December 1, 2012.
40. Jueckstock JK, Kaestner R, Mylonas I. Managing hyperemesis gravidarum: a multimodal challenge. BMC Med. 2010;8
41. Montouris G. Gabapentin exposure in human pregnancy: results from the Gabapentin Pregnancy Registry. Epilepsy Behav. 2003;4(3):310–317.
42. Park-Wyllie L, Mazzotta P, Pastuszak A, et al. Birth defects after maternal exposure to corticosteroids: prospective cohort study and meta-analysis of epidemiological studies. Teratology. 2000;62(6):385–392.
[loginform]