
 by Evangelia Bountouvi, MD, PhD; Melpomeni Giorgi, MD; Anna Papadopoulou, PhD; Kaj Blennow, PhD; Ingemar Björkhem, PhD; Maria Tsirouda, PhD; Spyridon Kanellakis, Msc, PhD; Andreas Fryganas, MD; Maria Spanou, MD; Ioanna Georgaki, MD; Sofia Asprogeraka, MD; and Argyrios Dinopoulos, PhD
by Evangelia Bountouvi, MD, PhD; Melpomeni Giorgi, MD; Anna Papadopoulou, PhD; Kaj Blennow, PhD; Ingemar Björkhem, PhD; Maria Tsirouda, PhD; Spyridon Kanellakis, Msc, PhD; Andreas Fryganas, MD; Maria Spanou, MD; Ioanna Georgaki, MD; Sofia Asprogeraka, MD; and Argyrios Dinopoulos, PhD
Drs. Bountouvi, Giorgi, Papadopoulou, Tsirouda, Fryganas, Spanou, Georgaki, Asprogeraka, and Dinopoulos are with the Third Department of Pediatrics, Athens University Medical School, at the University General Hospital “Attikon” in Athens, Greece. Dr. Blennow is with the Institute of Neuroscience and Physiology, Department of Psychiatry and Neurochemistry, at the Sahlgrenska Academy at the University of Gothenburg in Mölndal, Sweden, and the Clinical Neurochemistry Laboratory at Sahlgrenska University Hospital in Mölndal, Sweden. Dr. Björkhem is with the Department of Laboratory Medicine, Karolinska Institute, at the Karolinska University Hospital Huddinge in Huddinge, Sweden. Dr. Kanellakis is with the Department of Nutrition and Dietetics, Harokopio University, Kallithea, in Athens, Greece.
FUNDING: No funding was provided for this study.
DISCLOSURES: The author has no conflicts of interest relevant to the content of this article.
ABSTRACT: Niemann-Pick Type C disease (NPC) is a rare, incurable, autosomal-recessive, lysosomal storage disorder with protean and progressive neurovisceral manifestations characterized by accumulation of intracellular unesterified cholesterol. The investigational use of 2-hydroxypropyl-beta-cyclodextrin (HP-β-CD) in the treatment of NPC has shown promising results in improving life expectancy and reducing neurological damage in this patient population. This case report describes two children with the neurological form of NPC: a 5-year-old male patient in advanced stage of the disease and an 11-year-old female patient in moderately advanced stage. Despite treatment with the enzyme inhibitor, miglustat, both patients continued to exhibit severe neurodegeneration. High intrathecal (900mg) and low intravenous (350–500mg/kg) doses of HP-β-CD (Trappsol®Cyclo™) were administrated twice monthly to the patients in addition to miglustat therapy. The patients were monitored clinically as well as by imaging, laboratory, and biomarker (e.g., total tau protein [T-tau]; phosphorylated tau [P-tau]; neurofilament light [NFL], oxysterols) studies over a period of 16 to 22 months. The combination therapy of miglustat and HP-β-CD resulted in disease stabilization in both patients. The combination therapy demonstrated a good safety profile, and no adverse effects on hearing were observed. Additionally, CSF biomarkers appeared useful in monitoring neuronal damage. Large, randomized studies are needed to confirm these findings.
Keywords: Niemann-Pick type C disease (NPC), cyclodextrin, biomarkers, adverse events, total tau protein (T-tau), phosphorylated tau (P-tau), neurofilament light (NFL)
Innov Clin Neurosci. 2021;18(1–3):
Niemann-Pick type-C disease (NPC) is an incurable genetic neurovisceral disorder highlighted by excess endolysosomal accumulation, primarily of unesterified cholesterol. Clinical spectrum features a diverse constellation of visceral, psychiatric, and predominately neurological symptoms, leading to premature death. Miglustat (N-butyl-deoxynojirimycin), the only approved disease-modifying therapy, shows low efficacy in early-onset or advanced disease.1,2 Preclinical data and early-in-human evidence emerged by the investigational use of 2-hydroxypropyl-beta-cyclodextrin (HP-β-CD) is promising, reproducing its efficiency in decreasing neurological damage while also showing an acceptable safety profile, with hearing loss as the salient toxicity. Intrathecal (IT) outweighs intravenous (IV) delivery regarding neurological manifestations, while amelioration of visceral disease has been described via IV route. Two preparations are commercially available: Trappsol®Cyclo™ and VTS-270.3,4
Herein, we reported longitudinal data on safety and efficiency of semimonthly combined administration of high intrathecal and low intravenous dosages of HP-β-CD in two patients with NPC1.
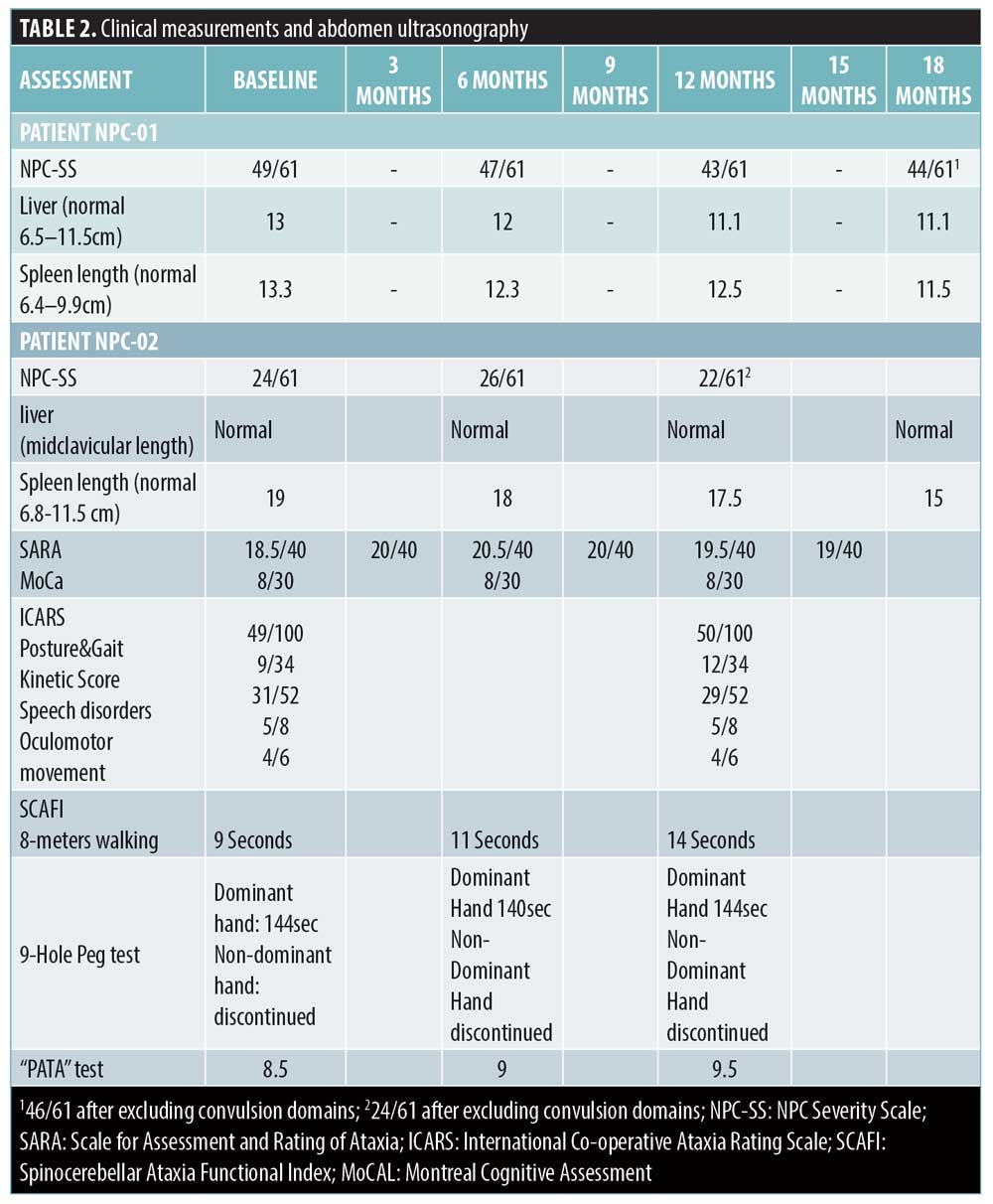
Case Report
NPC-01, a male patient with the early-infantile neurological form, was diagnosed when he was 3 months old, based on jaundice, hepatosplenomegaly, and early-onset hypotonia, via both the filipin test and genetic analysis of the NPC1 gene (c.2102dupA/[c.3265G>A Glu1089Lys]). Despite miglustat introduction at the age of 2 years, the patient’s condition rapidly deteriorated. He was evaluated by our team at the age of 5 years, and HP-β-CD was initiated. By that time, he was in advanced stage (National Institutes of Health NPC-Severity Scale [NPC-SS] 49/61);5 cachectic, bedridden, with protracted neurodegeneration, profound hearing loss, no verbal communication, vertical supranuclear gaze palsy (VSGP), rigidity, extensive skeletal deformities, intractable seizures (5–6 daily), along with profound hepato- and splenomegaly (liver midclavicular length 13cm [normal range 6.5–11.5], spleen length 13.3cm [normal range 6.4–9.9], as assessed by ultrasonography) and incipient interstitial lung disease. Besides miglustat, he had been treated with multiple antiepileptics (by that time Levetiracetam [LEV] and Valproate acid [VPA]).
NPC-02 was a female patient with the late infantile neurological form. Soon after birth, she had been evaluated for hepatosplenomegaly and persistently elevated liver enzymes, which were attributed to congenital CMV infection. Subsequently, her development was within normal range up to the age of 3 years, when she presented with mild ataxia followed by VSGP. NPC diagnosis was established by the filipin test and NPC1 molecular analysis at the age of 5 years (c.3394G>C, Ala1132Pro homozygosity). Substrate reduction therapy with miglustat was introduced at the age of 6 years with suboptimal success, since the patient progressively deteriorated. Notably, the last year before evaluation by our team, a significant decline in the patient’s condition was reported by the parents; the latter led them to refer to our department. Thus, at the age of 11 years, she was assessed and enrolled in the HP-β-CD protocol. She was in a moderate advanced stage (NPC-SS 24/61), exhibiting ataxic gait, slurring but understandable speech, mild dysphagia with no aspirations, VSGP, high-frequency hearing loss, gelastic cataplexy, narcolepsy, intractable epilepsy, cognitive impairment, liver size at upper normal limits, and splenomegaly (spleen length 19cm [normal range 6.8–11.5], as assessed by ultrasonography). Her regimen included miglustat and antiepileptics (LEV, VPA).
Written consent was obtained for both patients. The protocol of the study was a priori developed based on literature review and approved by the Greek National Drug Organization and the Institutional Review Board.4,6 A standardized formulation (Trappsol®Cyclo™) was semimonthly employed via lumbar puncture with gradual escalation (200–900mg). Maximum dose and intervals of IT route were determined based on the good safety profile of HP-β-CD and on evidence in NPC feline.4 After two hours, Trappsol was slowly IV infused (6–8 hours) with final escalation to 350–500mg/kg. Nearly 6–8mL of CSF as well as blood and urine were collected prior to HP-β-CD infusion. In both patients, the miglustat therapy was retained and the anticonvulsive regimen was modified. Disease progress was thoroughly assessed by neurological/pediatric examinations and objective measures (Supplemental Table 1). Biomarkers analysis in CSF included total-tau protein (T-tau) and phosphorylated-tau (P-tau) (reflecting neurodegeneration), neurofilament light protein (NFL) (reflecting damage to long myelinated subcortical axons (7)) and 24(S)- and 27-hydroxycholysterol. T-tau concentration was measured using ELISA, P-tau -phosphorylated at threonine 181 – using sandwich – ELISA [InnotesthTAU-Ag and InnotestPhospho-Tau (181P); Fujirebio Europe, Ghent,Belgium, respectively] and NFL using a novel ELISA-method based on monoclonal antibodies.8 Oxysterols levels were determined by mass spectrometry.9 NPC-01 and NPC-02 were monitored over a period of 22 and 16 months, respectively. Adverse events were categorized in accordance with Common Terminology Criteria for Adverse Events (AEs-CTCAE Version 4).
Results and Discussion
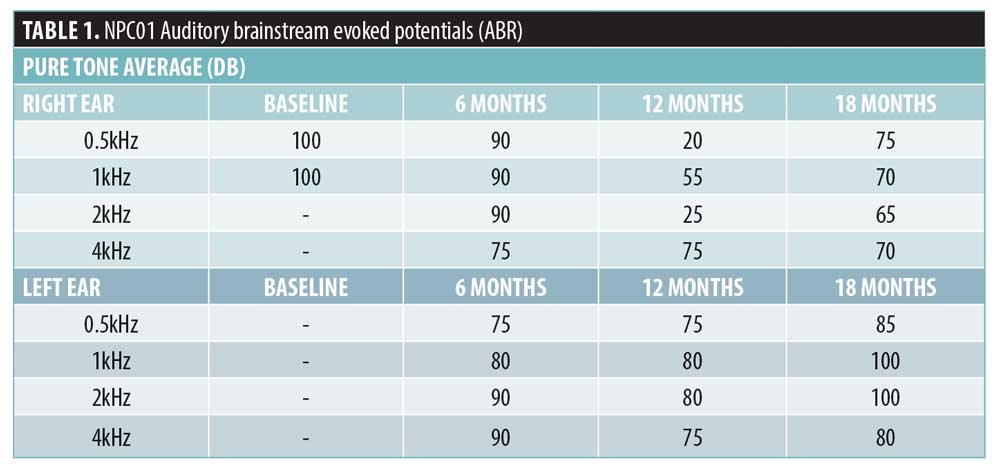
NPC-01 received, over a period of 22 months, 43 HP-β-CD infusions, achieving maximum dose after the seventh infusion. NPC-02 received, over a period of 16 months, 31 HP-β-CD infusions, achieving maximum dose after the fourth infusion. The only mild AEs that were noted in both cases (i.e., fever, shudder, dizziness/ataxia, and seizures, which are known to be associated with higher IT doses)10,11 gradually attenuated (Supplemental Table 2). Auditory dysfunction has been commonly reported; remarkably, this was the case in all 14 patients in the recent Phase 1 and 2 clinical trial with VTS-20.10,11 Intriguingly, NPC-01 showed improvement; initial profound hearing loss was enhanced to moderate at Month 12 with some deterioration at Month 18 (Table 1). NPC-02 displayed mild high frequency sensorineural hearing loss unchanged from baseline (Pure tone average >15dB in 4-8kHz). In our probands, the beneficial effect of cyclodextin counteracts its toxicity on cochlea; this could be attributed to the combination of dosing/route implemented and/or intrapersonal variability.
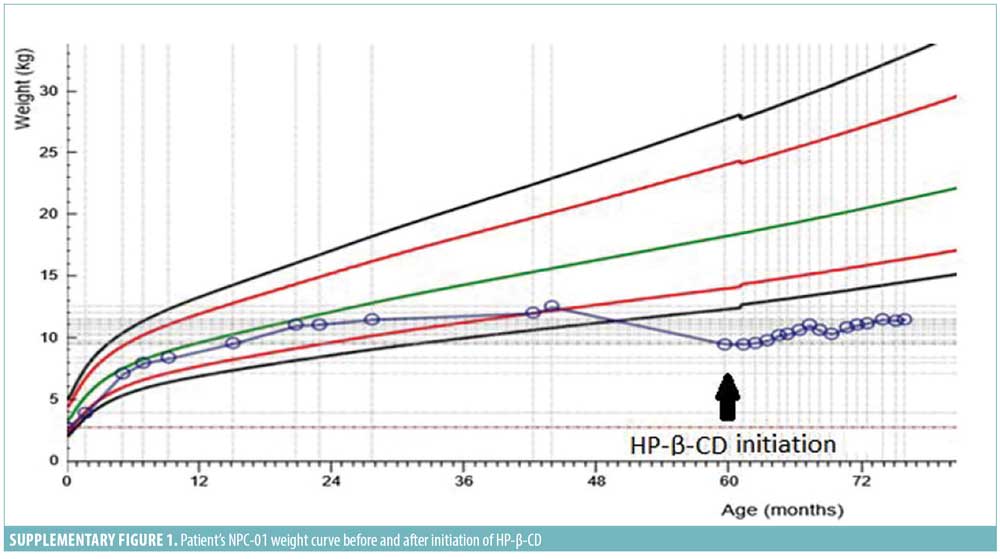
Overall, detailed clinical, imaging, and biochemical follow-up for almost two years converge into a rather stabilized neurodegenerative phenotype in both patients via IT route, against the natural deteriorating course of the disease. 5 After excluding convulsion domains, which required the exploitation of additional therapy, NPC-SS decreased three points for NPC-01 at Month 18, mirroring improvement in hearing and swallowing, and was stable for NPC-02 at Month 12 compared to pretreatment. NPC-01’s initial swallowing evaluation revealed acceptable pharyngeal phase with oral apraxia, which improved at Month 18. Indeed, less coughing while eating was reported, thus further weight loss was prevented; notably, 3kg were gained (Supplementary Figure 1). MRS revealed mild improvement with increased N-acetylaspartate/creatine and decreased choline/creatine ratio at centrum semioval, demonstrating reduction in neuronal and myelin destruction. No substantial differentiation was depicted in the electroencephalograms (EEGs). Finally, family and physiotherapists reported amelioration in patients’ management, rigidity, sleeping hours, and general mood.
NPC-02’s NPC-SS were stabilized, despite the steady increase during the last two years before HP-β-CD initiation (7-point increment). NPC-02 had already been suffering from seizures refractory to anticonvulsants before study enrollment. Convulsions control was achieved under Valproate acid and Perampanel nearly seven months after HP-β-CD initiation, and episodes of cataplexy and narcolepsy became more apparent but responded to Fluoxetine. This course was also depicted in an EEG performed at Month 12, which demonstrated improvement of epileptic activity and profound impairment of alertness stage compared with baseline. As aforementioned, since additional drugs were administered, improvement in convulsions domains were not considered for evaluation of HP-β-CD efficiency. Thorough neurological assessment mainly revealed disease stabilization, with the only differentiation being a slight deterioration in posture/gait and balance, depicted at SARA, ICARS, and 8m-timed walking test but not reflected in the NPC-SS. The rest of the domains assessed remained stable (no changes in 9-HPT, ‘PATA’ rate, MoCa; Table 2). Swallowing function was assessed at baseline as mild delay of oral phase with no obvious difficulties or aspirations and remained unchanged at Month 12. Brain MRI/MRS (spectroscopy findings at lower normal range) were also invariable at reevaluation. Overall disease progression was stabilized, showing only a slight worsening of gait/posture not reflected in NPC-SS (Table 2).
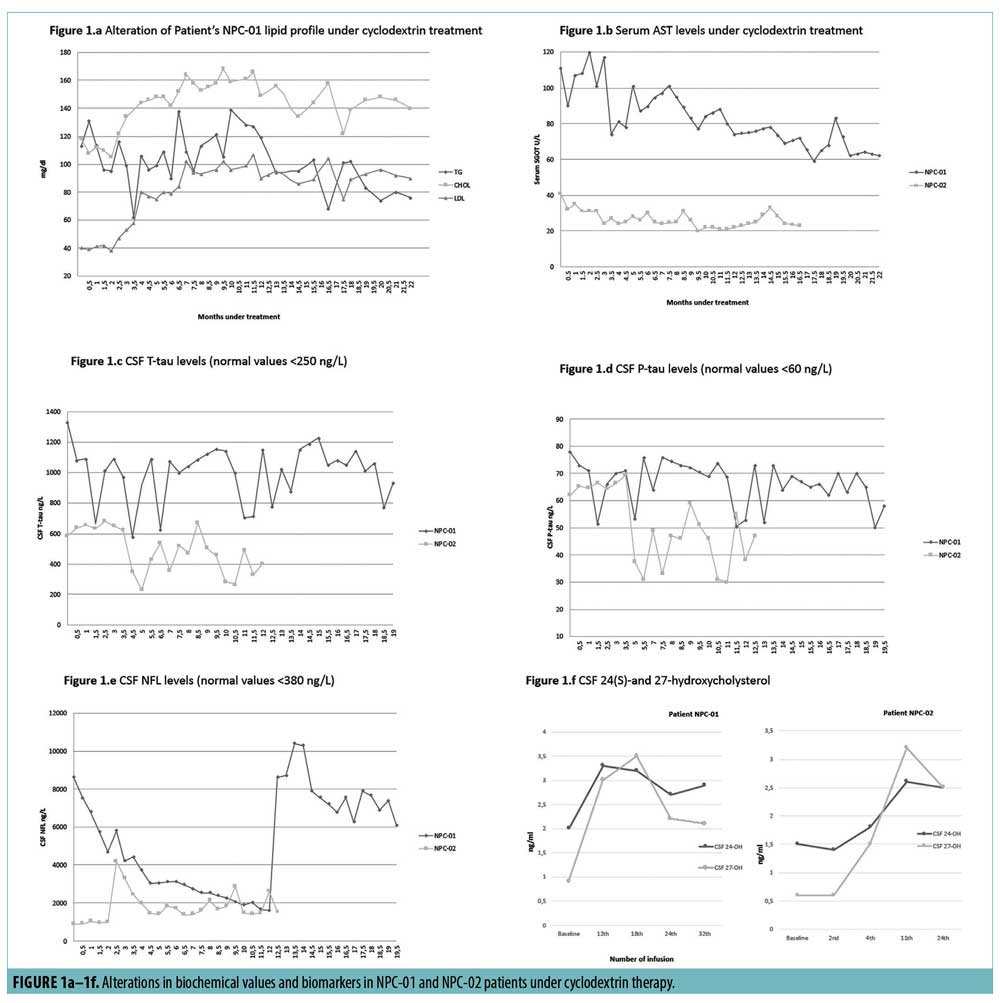
HP-β-CD was IV administrated mainly at 2–2.5g/kg twice/thrice weekly.3 We hypothesized that lower doses might be efficient in visceral manifestations, as neurodegeneration was faced IT. The therapeutic schedule was within the safety profile, contrary to other reports.3 Some improvements in visceral disease were observed, indicating redistribution of stored cholesterol. In particular, slight gradual reduction in NPC-01’s hepatosplenomegaly and NPC-02’s splenomegaly (Table 2) as well as mild progressive decline of AST levels in both patients were demonstrated. In NPC-01, alkaline phosphatase was incremented to normal values (from 70U/Lto~140U/L), low-density lipoprotein (LDL) levels were substantial increased, total cholesterol tended to raise and triglycerides fell to lower levels (Figure 1a–b). Remaining biochemical parameters were virtually stable. Noteworthy, outcomes of visceral disease were similar to those reported in two patients, received much higher and repeated IV doses.12,13
Changes in CSF T-tau, P-tau, NFL, 24(S)- and 27-hydroxycholysterol are shown in Figure 1c–f. T-tau and P-tau concentrations were substantially elevated at baseline and declined under HP-β-CD therapy (more in NPC-02), in line with data in three other patients with NPC under miglustat.14 CSF NFL is proposed as a downstream biomarker for neurodegeneration. NFL levels followed a regressive slope in NPC-01, interrupted by a burst at Month 12, possibly linked to a febrile viral infection. In NPC-02, NFL-levels were variable, probably reflecting a more active disease. Augmentations of NFL-levels were observed after either lowering/skipping a dose due to infections or seizures/cataplexy intensification. Potential explanations could be that a) 900mg semimonthly was more suitable IT-dose for our patients or NFL-levels might be influenced by infections, b) peaks might reflect a modified disease progression from linear to occasionally bursting, and c) clinical severity might show some correlation to NFL-levels. Overall, data converges into a rather positive impact of HP-β-CD on axonal degeneration and neurodegenerative progress. Although CSF 24(S)hydroxycholysterol and 27-hydroxycholysterol levels were measured pre-dosing, they were found to be stabilized to a rather higher level, compared to baseline, which has been commonly reported in patients with NPC and animal models postdosing.6,10 Twenty-four (S)hydroxycholysterol is indicative of the target engagement of HP-β-CD, while 27-hydroxycholysterol increment might reflect an impact of HP-β-CD on blood-brain barrier.15
Conclusion
In conclusion, our work is the first report assessing such an HP-β-CD dosage combination, as well as presenting thorough biomarker analyses of CSF-neurodegeneration markers. The drug combination applied, targeted directly at both the neurological (IT route) and visceral (IV route) aspects of the disorder, led to rather stabilizing the disease for both patients within good safety profile. Intriguingly, no adverse effect on hearing was observed. These results along with the role of CSF-biomarkers of neuronal damage in monitoring neurodegeneration in NPC justify further investigation.
Acknowledments
The authors are indebted to the patients and their families for their collaboration with this study. Dr. Ingemar Björkhem has received a grant from “The Stockholm County Council (ALF).”
References
- Vanier MT. Niemann-Pick disease Type C. Orphanet J Rare Dis. 2010;5:16.
- Pineda M, Walterfang M, Patterson MC. Miglustat in Niemann-Pick disease Type C patients: a review. Orphanet J Rare Dis. 2018;13(1):140.
- Megías-Vericat JE, Company-Albir MJ, García-Robles AA, Poveda JL. Cyclodextrin – a versatile ingredient. London, UK: IntechOpen; 2018:95–117.
- Vite CH, Bagel JH, Swain GP, et al. Intracisternal cyclodextrin prevents cerebellar dysfunction and Purkinje cell death in feline Niemann-Pick type C1 disease. Sci Transl Med. 2015;7(276):276ra26.
- Yanjanin NM, Velez JI, Gropman A, et al. Linear clinical progression, independent of age of onset, in Niemann-Pick disease, type C. Am J Med Genet B Neuropsychiatr Genet. 2010;153B(1):132–140.
- Maarup TJ, Chen AH, Porter FD, et al. Intrathecal 2-hydroxypropyl-beta-cyclodextrin in a single patient with Niemann-Pick C1. Mol Genet Metab.
2015;116(1–2):75–79. - Blennow K, Hampel H, Weiner M, Zetterberg H. Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat Rev Neurol. 2010;6(3):131–144.
- Gaetani L, Hoglund K, Parnetti L, et al. A new enzyme-linked immunosorbent assay for neurofilament light in cerebrospinal fluid: analytical validation and clinical evaluation. Alzheimers Res Ther. 2018;10(1):8.
- Dzeletovic S, Breuer O, Lund E, Diczfalusy U. Determination of cholesterol oxidation products in human plasma by isotope dilution-mass spectrometry. Anal Biochem. 1995;225(1):73–80.
- Ory DS, Ottinger EA, Farhat NY, et al. Intrathecal 2-hydroxypropyl-beta-cyclodextrin decreases neurological disease progression in Niemann-Pick disease, type C1: a non-randomised, open-label, phase 1-2 trial. Lancet. 2017;390(10104):1758–1768.
- Berry-Kravis E, Chin J, Hoffmann A, et al. Long-term treatment of Niemann-Pick type C1 disease with intrathecal 2-hydroxypropyl-beta-cyclodextrin. Pediatr Neurol. 2018;80:24–34.
- Megias-Vericat JE, Garcia-Robles A, Company-Albir MJ, et al. Early experience with compassionate use of 2 hydroxypropyl-beta-cyclodextrin for Niemann-Pick type C disease: review of initial published cases. Neurol Sci. 2017;38(5):727–743.
- Chien YH, Shieh YD, Yang CY, et al. Lung toxicity of hydroxypropyl-beta-cyclodextrin infusion. Mol Genet Metab. 2013;109(2):231–232.
- Mattsson N, Zetterberg H, Bianconi S, et al. Miglustat treatment may reduce cerebrospinal fluid levels of the axonal degeneration marker tau in niemann-pick type C. JIMD Rep. 2012;3:45–52.
- Leoni V, Masterman T, Patel P, et al. Side chain oxidized oxysterols in cerebrospinal fluid and the integrity of blood-brain and blood-cerebrospinal fluid barriers. J Lipid Res. 2003;44(4):793–799.