
 by Monica Javidnia, PhD; Mark Frasier, PhD; Ira Shoulson, MD; Ibrahim Turkoz, PhD; and Kumar Budur, MD, MS
by Monica Javidnia, PhD; Mark Frasier, PhD; Ira Shoulson, MD; Ibrahim Turkoz, PhD; and Kumar Budur, MD, MS
Drs. Javidnia and Shoulson are with the Center for Health + Technology and the Department of Neurology, University of Rochester Medical Center in Rochester, New York. Dr. Shoulson is also with Grey Matter Technologies in Sarasota, Florida. Dr. Frasier is with the Michael J. Fox Foundation for Parkinson’s Research in New York, New York. Dr. Turkoz is with Janssen Research & Development in Titusville, New Jersey. Dr. Budur is with AbbVie in Lake Bluff, Illinois.
FUNDING: No funding was provided.
DISCLOSURES: The authors have no conflicts of interest relevant to the content of this article.
ABSTRACT: The International Society for Central Nervous System Clinical Trials and Methodology (ISCTM) partnered with the Michael J. Fox Foundation for Parkinson’s Research to hold a joint session on innovation in Parkinson’s disease research at the ISCTM 14th Annual Scientific Meeting held February 20 to 22, 2018 in Washington, D.C. The session focused on (1) biomarkers and outcomes measures in Parkinson’s disease clinical trials; 2) clinical trial designs, outcomes, and statistical approaches; and 3) the path forward. This paper aims to summarize key takeaways from the session presenters, panelists, and audience members.
KEYWORDS: Parkinson’s disease, prodromal, RBD, disease progression, disease modification
Innov Clin Neurosci. 2020;17(14–19)
It has been just over 200 years since James Parkinson wrote “An Essay on the Shaking Palsy,” describing what was later named “Parkinson’s disease” (PD) by Jean-Martin Charcot. When discussing a cure, Parkinson wrote, “…remedial means might be employed with success: and even, if unfortunately deferred to a later period, they might then arrest the farther progress of the disease, although the removing of the effects already produced might be hardly to be expected.”
Since then, there have been numerous advances in therapeutics, mostly that treat the motor complications of the disease, yet there remains an urgent need for treatment options which can slow, halt, or reverse PD progression. A recent analysis projects over one million people in the United States will have PD by 2020 and nearly two million will be afflicted by 2060.1 To address this looming pandemic, the International Society for Central Nervous System Clinical Trials and Methodology (ISCTM), in partnership with the Michael J. Fox Foundation for Parkinson’s Research (MJFF), held a joint session on Innovative Approaches for Slowing Disease Progression in Parkinson’s Disease on February 22, 2018 at the 14th Annual ISCTM Scientific Meeting in Washington, D.C.
There are several problems that the PD research and patient community face, including the heterogeneous and multisymptomatic nature of the disease, the amount of neurodegeneration prior to motor symptom manifestation, and numerous failures in clinical trials. PD is a complex neurodegenerative disorder, encompassing numerous motor and nonmotor symptoms.2 Diagnosis is associated with the onset of motor features, including tremor, bradykinesia, postural instability, and rigidity. However, nonmotor symptoms such as constipation, olfactory dysfunction (i.e., hyposmia, anosmia), sleep disturbances, and mood disorders (e.g., depression, anxiety) can precede motor symptoms by years and potentially decades.3 Nonmotor symptoms can persist throughout the course of the disease and grow to include cognitive dysfunction (e.g., PD mild cognitive impairment,4 PD dementia5), pain, hypotension, genitourinary dysfunction, psychosis (hallucinations, delusions), excessive daytime sleepiness, apathy, and other symptoms.6 Collectively, the burden of PD symptoms have a substantial impact on quality of life and disability.7–10
This session aimed to share breakthroughs; stimulate meaningful and collaborative discussion; and build consensus in the field on disease targets, clinical trial design, and regulatory considerations in PD therapeutic development. The following report covers the three general areas of the session and is intended to be a summary of the key findings, suggestions, and challenges identified by speakers, panelists, and audience members. For clarity, the following terms are defined below.
- Disease-modifying: halting or slowing disease progression by favorably modifying underlying disease mechanisms
- Preclinical: Prior to the manifestation of symptoms; does not include animal studies (nonclinical)
- Prodromal: Prior to the diagnosis of Parkinson’s disease; early, nonmotor symptoms are present
Part I: Biomarkers and Outcomes Measures in PD Clinical Trials
Targeting preclinical and prodromal PD. At least half of patients with PD will begin dopaminergic treatment within 12 months of their diagnosis, creating a challenge for clinical research. These drugs are not just highly effective at ameliorating motor symptoms but are also given when the pathology is fairly advanced in the brain.11 Prodromal PD, the period of time during which subtle signs are present but clinical PD is not yet manifest, is estimated to be at least 10 years long, providing a large window of opportunity to intervene. However, it also poses a unique challenge on the optimal time for intervention because current prodromal symptoms or putative biomarkers of disease prediction/progression do not predict the age of onset of clinical symptoms. Unlike Alzheimer’s disease (AD), wherein the prodromal phase is generally considered to involve mild cognitive impairment, PD’s prodromal phase comprises multiple symptoms, many of which are nonspecific and not infrequent in the general population. Therefore, it is crucial to understand and accurately identify the prodromal phase of PD.
In 2015, the Movement Disorder Society (MDS) published criteria and methodology for using clinical risk markers to calculate the probability of prodromal PD.12 Clinical markers include constipation and hyposmia, with the most compelling predictor being the presence or history of rapid eye movement sleep behavior disorder (RBD), an increasingly recognized condition involving dream enactment with dramatic dreaming and loss of motor and behavioral inhibition. In a study following 174 patients with RBD, the risk for being diagnosed with a defined neurodegenerative disease was 90.9 percent at 14 years post-RBD diagnosis,13 with a strong association of RBD with synucleinopathies. Thus, clinical trials could target patients with RBD and measure the time to develop features of PD or dementia with Lewy bodies (DLB).
There are practical complexities to consider when testing interventions in early PD and prodromal populations, such as the safety concerns of treating “healthy” individuals. Prior to clinical trials in prodromal or preclinical PD, several logistical issues should be addressed, including:
- With the long presymptomatic period, how long should the duration of the trial be?
- What clinical and biomarker endpoints should be measured and how much follow-up time would be required?
- What are the safety concerns in administering potentially long-term experimental therapeutics to individuals who have not yet developed or who might never develop PD?
- Will early intervention exert a clinically significant effect?
- How do you identify and reach out to people who are unimpaired and get them to volunteer for research?
The Parkinson’s Associated Risk Study (PARS) incorporated a groundbreaking approach to finding potential prodromal patients with PD. This study identified individuals over the age of 60 years without clinical features of a neurodegenerative disorder, then mailed an eligibility questionnaire and “smell test” to be completed at home.14–18 Another method to identify potential prodromal individuals is by using the REM Sleep Behavior Disorder Single Question Screen (RBD1Q).19 A test case in a community newspaper found that, of 111 respondents to a newspaper ad containing the RBD1Q, 19 (17%) had RBD proven on polysomnography and 80 percent of these met MDS criteria for prodromal PD.20 In spite of these successes, there are potential issues of outreach and recruitment that have not been adequately articulated and discussed.
Utility of biomarkers in PD. Tests involving tissue, fluid, and imaging markers have the potential for increasing the accuracy of PD diagnosis, improving study design, and tracking disease progression. One such example is dopamine transporter (DAT) neuroimaging, which allows researchers to differentiate patients with PD from individuals initially diagnosed as having PD but who instead have scans without evidence of dopaminergic deficit (SWEDD) (follow-up of these individuals has shown that they generally do not evolve to have typical PD). The Critical Path Institute’s Critical Path for Parkinson’s Consortium (CPP) was able to glean a qualification opinion from the European Medicines Agency (EMA) for the use of baseline DAT neuroimaging for patient selection in early PD clinical trials in 201821 as well a Letter of Support from the United States Food and Drug Administration (FDA) and the EMA. The EMA Committee for Medicinal Products for Human Use (CHMP) is able to issue a qualification opinion on the acceptability of the specific use of a method for research and development. Similarly, the FDA Letter of Support Initiative entails publicly posted documents that indicate a regulatory agency’s views on a biomarker’s potential and encourages further studies and sharing of data. It is important to note that the utility of a biomarker in research and clinical practice will depend not only on its value, accuracy, and precision but also the cost and invasiveness of the procedure. Trial enrichment requiring a lumbar puncture, e.g., might negatively affect recruitment while imaging markers, though less invasive, might be more expensive.
Currently, clinical markers are the most reliable means for diagnosing prodromal PD, though there is growing evidence supporting the use of imaging and tissue markers. Phosphorylated alpha-synuclein (pS129) has been detected in submandibular gland and dermal biopsies from individuals with RBD.22–24 In all three studies, pS129 was not detected in healthy controls. Other nonclinical markers with diagnostic potential for prodromal PD include nigral ultrasound25 and magnetic resonance imaging.26, 27
Data accrual, standardization and sharing. Developing standards for data collection is crucial, allowing for comparisons and meta-analyses to be made across data repositories. Efforts toward this are in progress by groups such as the CPP consortium, which has already collected and standardized world-wide PD patient data into a single, unified database. Additionally, through MJFF’s Linked Efforts to Accelerate Parkinson’s Solutions, alpha-synuclein assay standardization studies have compared assay platforms in multilaboratory studies.28
A major initiative in the PD field is the Parkinson’s Progression Markers Initiative (PPMI). Sponsored by MJFF, PPMI longitudinally collects comprehensive clinical, imaging, and biofluid data from healthy, prodromal, SWEDD, newly diagnosed and drug-naïve PD, and PD-related genetic mutation–carrying (SNCA, LRRK2GB, or GBA) research participants. The rationale behind PPMI was to develop a biomarker-focused cohort as well as conduct the standardized acquisition of data and data sharing. Roche and Verily are expected to introduce wearables to the study, allowing for regular assessments and monitoring of gait, mobility, balance, and other measures without the need for additional clinic visits. Such technologies can provide more data than would be collected in-person, and it is important to understand how to store, analyze, and extract meaningful results that can be used to change the landscape of PD research and care.
Potential targets for disease-modifying therapy. PD is complicated from many perspectives, one being that there are multiple genes that account for the same or similar clinical manifestations. There are rare mutations that have a large clinical effect (e.g., mutations in LRRK2 G2019S) and common genetic variants (e.g., variants in MAPT, SNCA, and many other genes) that exert a relatively small effect size, but which can prove detrimental when combined. Four gene-derived targets—SNCA, LRRK2, GBA1, and HLA—were previously highlighted by Standaert (Table 1), with targeted therapies recently reviewed by Sardi et al.29 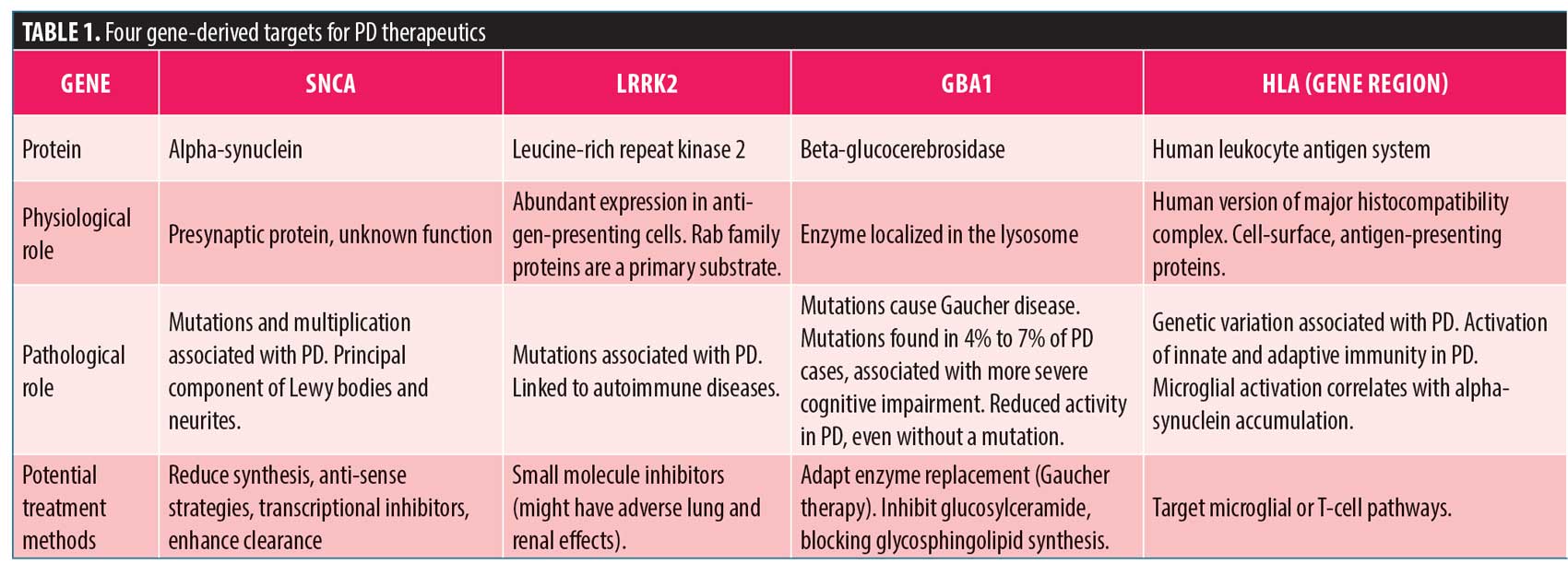
While PD is a heterogeneous disease, neuroinflammation and alpha-synuclein accumulation occur in the majority of PD cases. Thus, interventions targeting these processes might have the broadest application. There remain questions regarding the underlying pathology that are left unanswered, including:
- What are the pathological forms of alpha-synuclein?
- Is the accumulation of alpha-synuclein in Lewy bodies protective or pathological?
- Do processes such as neuroinflammation, mitochondrial dysfunction, and failures in protein clearance lead to alpha-synuclein pathology or are they secondary to or independent of it?
Overall, there is a need to enhance the basic understanding of disease mechanisms to ensure therapeutic efforts are directed toward the appropriate targets.
Part II: Clinical Trial Designs, Outcomes, and Statistical Approaches
Disease modification versus disease progression. Disease modification is defined as a beneficial outcome characterized by slowing, halting, or reversing of the progression of disease (as reviewed by Lang et al.30). In contrast, disease progression is understood as a worsening of a disease in terms of symptom severity, underlying pathology, or outcome. Part I of this report described potential targets for disease-modifying therapies, though questions still linger regarding the roles of specific alpha-synuclein conformations and Lewy bodies in PD and whether we know enough about the disease to proceed with costly and time-consuming trials. This raises the question of whether interventions should aim to modify the disease or alter disease progression. Slowing disease progression or delaying a disability outcome could and should be a significant advance and might not be considered as modifying the course of the disease. The type of experimental intervention and stage at which it is anticipated to be implanted will dictate the study design. Delayed-start designs, e.g., assume a homogeneous disease course and demand rigorous statistical methods to address limitations encountered during implementation. To address these limitations, we need to utilize improved study designs.
Adaptive designs. The PhRMA Working Group on adaptive clinical trial designs published a white paper in 2006 addressing various aspects of adaptive designs and recommendations from the group.31 The concept of adaptive designs can be intimidating as the trial design might be changed based on data accrued during the study. It is imperative these adaptations are made by design, without undermining the integrity of the trial and with prespecified rules. There are several adaptive designs that could be utilized in a study,32 some of which were described by Coffey (Table 2). Importantly, by cleaning and querying the data as they are generated, researchers are likely to identify both effective treatments more efficiently and “duds” earlier in the process. That is not to say that adaptive designs should be used universally, as they are not necessarily better than standard methods and might have minimal benefit given the increase in “up-front” planning and ongoing adaptations required. Properly planned adaptive designs might lead to more efficient trials that require less time and fewer resources than traditional designs. However, adaptive designs are not always “better”—the time required to perform simulations to justify the design might offset any “time” saved by the adaptations. Researchers should utilize adaptive designs only when necessary. It is also important to be clear that an adaptive design cannot change the answer regarding the effectiveness of a particular treatment. An adaptive design, i.e., cannot make a drug more effective. Rather, one of the biggest benefits of adaptive designs is the ability to identify ineffective treatments in a timelier manner.

Network of Excellence in Neuroscience Clinical Trials (NeuroNEXT)—a novel initiative. Though the statistical groundwork for adaptive designs can be developed, logistical barriers make it difficult to implement. To address this issue, there has been a movement in industry toward the creation of in-house teams primarily responsible for modeling clinical trials to assess various design options. Greater barriers exist for implementing this type of infrastructure within the publicly funded environment since grant applications require these complex simulations to be conducted before the design is finalized—i.e., before grant submission. Few grant-funded mechanisms exist to support these complex simulations. Consequently, there is a growing divide between the practicality and feasibility of conducting adaptive designs in industry versus academia. One initiative to address the shortcomings in academic clinical research is NeuroNEXT. This National Institute of Neurological Disorders and Stroke–funded network is designed to facilitate and conduct exploratory phase II neurological disease trials initiated by academia, industry, and foundations. The network utilizes a central institutional review board of record and Master Clinical Trial Agreements made with the coordination centers and sites. Proposals are evaluated for their network feasibility, mission relevance, and institute priority. Approved proposals are then referred to protocol working groups, consisting of experienced trial design staff, for further development and peer-reviewed applications for funding. The network infrastructure support for such design activities dramatically increases the feasibility for using more novel trial designs in academic clinical trials. Other National Institutes of Health–associated clinical trials networks include the Neuroprotection Exploratory Trial in Parkinson’s Disease (NET-PD), Alzheimer’s Disease Cooperative Study (ADCS), Neurological Emergencies Treatment Trial (NETT), and StrokeNet.
Part III: The Path Forward and Concluding Remarks
Clinical trial outcomes. The MDS-sponsored Unified Parkinson’s Disease Rating Scale (MDS-UPDRS) is the current gold standard for PD symptom assessment. This four-part scale covers key aspects of PD and includes other measures such as the Schwab and England Activities of Daily Living and Hoehn and Yahr scales. The slope for decline in UPDRS impairment scores is steeper earlier in the disease,33 due largely to the sensitivity of the scale for early impairment and the concomitant introduction of dopaminergic therapies.
Our current view of PD therapeutic efficacy is based on examining clinical endpoints using categorical scales, as rated by patients, investigators, or both. Many clinical trials utilize the MDS-UPDRS, particularly part III, as a primary endpoint for efficacy. Other trials such as DATATOP34 used “time to an important clinical event” (e.g., disability requiring dopaminergic therapy). A problem not unique to PD is that the clinical scales and “time to event” might not correlate with quality of life (QoL). Thus, long-term disability and QoL should be considered relevant endpoints for a study. Perhaps the best method is to include multiple endpoints and demonstrate efficacy in a number of meaningful aspects of the disease such as favorable changes in symptoms, functional capacity, and biology.
Potential therapeutics for PD can be grouped by one or more of the following classifications: (1) symptomatic or compensatory; (2) restorative (e.g., increasing dopaminergic neurons); and (3) protective or rescue (i.e., interfering with the cause of neurodegeneration). Thus, trials must be designed to utilize the appropriate biomarkers for a given intervention and study population.35 PD is more than a motor disease to be treated with motor symptoms targeting drugs and assessed on a motor scale. Therefore, clinical trial designs should address nonmotor features (e.g., cognition, sleep, fatigue, mood, constipation, pain) in addition to motor symptoms. It is important to incorporate innovative approaches for trial design (e.g., patient selection, trial format, and outcome measures) and to understand the benefits and limitations of each method. More recent observational studies like PPMI, e.g., can inform the calculation of sample-size estimates, followed by an internal pilot design for sample size re-estimation.
Lessons learned from historical data and other diseases. The analysis of historical clinical trial data is key to understanding the strengths and weaknesses of different approaches within the context of their study population and intervention and for providing measurements of disease progression or placebo effects to help improve trial efficiency. It is worth revisiting why seemingly promising trials might have failed and whether subgroup analysis might have shown signs of efficacy or futility. Historical and observational data might also help in developing disease modeling and simulation tools and might also aid regulatory agencies with understanding the assumptions and limitations of a proposed trial design. One example is the AD clinical trial simulation tool endorsed by the FDA and EMA developed by the Critical Path Institute’s CAMD consortium, now termed the Critical Path for Alzheimer’s Disease (CPAD), longitudinally modeling changes in cognition.36
It is important to examine other diseases in considering innovation for PD research, as lessons learned in cancer and AD research provide valuable groundwork for the study of PD. Success in research would require standardized definitions of disease staging and heterogeneity. To this end, the Alzheimer’s Association and National Institute of Aging have collaborated to propose a hypothetical diagnostic framework for research purposes only that uses amyloid beta, phosphorylated-tau, and total tau measurements.37,38 Furthermore, the FDA issued a draft guidance for drug development in early AD that provides recommendations for staging of patients with AD and outcome measures for the earlier stages. These guidelines provide groundwork for researchers to engage regulatory agencies and develop similar standards for PD.
In cancer research, the Investigation of Serial Studies to Predict Your Therapeutic Response with Imaging and Molecular Analysis (I-SPY) trial is in the second phase of a novel clinical trial design for patients with early-stage breast cancer. I-SPY 2 is a platform trial with up to 12 treatment arms being studied in participants stratified by standard biomarkers. This combined “basket” and “umbrella” design is intended to determine which drugs are effective and also which patients with specific disease subtypes might benefit. In addition to standard biomarkers, I-SPY 2 assesses qualifying and exploratory biomarkers. A recent viewpoint describes the potential benefits of adapting the I-SPY 2 method for AD trials.39 This model could also be applied to PD to improve study efficiency. Moreover, similar to in cancer and human immunodeficiency virus, it is likely that a successful disease-modifying therapy for PD will need to be combinatorial, perhaps targeting both alpha-synuclein and inflammation as well as other biological targets, including mitochondrial therapeutics currently under investigation that might also be vital for slowing disease progression.
Concluding remarks. The last ISCTM and MJFF joint session was in 2007. Karl Kieburtz closed the session by highlighting progress made in the PD field during this time. One can argue that progress has been slow for many patients who suffer from PD and the caregivers spending countless hours caring for these individuals. However, within this time frame, there have been countless breakthroughs in PD. These include but are not limited to new drug and device approvals; the establishment of registries with over 1,000 patients with biomarker data freely accessible to researchers worldwide; and a wide array of new tools for imaging and genetic analysis. While there are still many unknowns in PD research, we are regularly learning more about risk factors and pathological processes, and many ongoing clinical trials are targeting the underlying disease mechanisms. We have made progress on new trial designs and on building infrastructure to implement them. There are now diagnostic tools for early and prodromal PD, with more sensitive measures under investigation.
In order to accelerate the progress in developing better interventions for PD, a collective effort is required, such as the Linked Clinical Trials (LCT) Initiative,40,41 which involves a worldwide clinical development program currently considering over 40 distinct biological targets, each with disease-modifying potential for patients with PD. More frequent meetings and discussions that bring together regulators, patients, industry, and academia are needed to address specific challenges facing the PD landscape. Regulatory engagement and input are critical to provide draft guidance to the research community around common standards and expectations when developing new treatments for PD. Data from human studies and trials should be shared through partnerships to increase our understanding of the disease and to support sponsors and regulators in designing better trials. Consortia like the CPP provide a venue for the needs of stakeholders (e.g., patients, industry, regulators) to be addressed, and these efforts should be expanded. The authors applaud the ISCTM and the MJFF for bringing together an enthusiastic and dynamic group of individuals to tackle these challenges. We encourage more frequent and open discussions involving PD stakeholders to tackle the barriers and accelerate the development of more effective treatments for patients.
Acknowledgments
The authors would like to thank the ISCTM and MJFF for hosting this joint session and the speakers Anthony Lang, Ron Postuma, David Standaert, Ken Marek, Karl Kieburtz, Christopher Coffey, and Pierre Tariot, and the panelists, Christopher Leptak, David Stamler, Diane Stephenson, Larry Alphs, Ravi Anand, Atul Bhattaram, Kun Jin, Michael Gold, Mark Frasier, Daniel Keene, and Gerald David Podskalny.
References
- Savica R, Grossardt BR, Rocca WA, Bower JH. Parkinson disease with and without Dementia: A prevalence study and future projections. Mov Disord. 2018;33(4):537–543.
- Kalia LV, Lang AE. Parkinson’s disease. Lancet. 2015;386(9996):896–912.
- Schapira AH, Tolosa E. Molecular and clinical prodrome of Parkinson disease: implications for treatment. Nat Rev Neurol. 2010;6(6):309–317.
- Litvan I, Goldman JG, Troster AI, et al. Diagnostic criteria for mild cognitive impairment in Parkinson’s disease: Movement Disorder Society Task Force guidelines. Mov Disord. 2012;27(3):349–356.
- Emre M, Aarsland D, Brown R, et al. Clinical diagnostic criteria for dementia associated with Parkinson’s disease. Mov Disord. 2007;22(12):1689–1707; quiz 837.
- Schapira AHV, Chaudhuri KR, Jenner P. Non-motor features of Parkinson disease. Nat Rev Neurosci. 2017;18(8):509.
- Barone P, Antonini A, Colosimo C, et al. The PRIAMO study: A multicenter assessment of nonmotor symptoms and their impact on quality of life in Parkinson’s disease. Mov Disord. 2009 Aug;24(11):1641–1649.
- Chapuis S, Ouchchane L, Metz O, et al. Impact of the motor complications of Parkinson’s disease on the quality of life. Mov Disord. 2005;20(2):224–230.
- Martinez-Martin P, Rodriguez-Blazquez C, Kurtis MM, et al. The impact of non-motor symptoms on health-related quality of life of patients with Parkinson’s disease. Mov Disord. 2011;26(3):399–406.
- Weintraub D, Moberg PJ, Duda JE, et al. Effect of psychiatric and other nonmotor symptoms on disability in Parkinson’s disease. J Am Geriatr Soc. 2004;52(5):784–788.
- Braak H, Del Tredici K, Rub U, et al. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24(2):197–211.
- Berg D, Postuma RB, Adler CH, et al. MDS research criteria for prodromal Parkinson’s disease. Mov Disord. 2015;30(12):1600–1611.
- Iranzo A, Fernandez-Arcos A, Tolosa E, et al. Neurodegenerative disorder risk in idiopathic REM sleep behavior disorder: study in 174 patients. PLoS One. 2014;9(2):e89741.
- Stern MB, Siderowf A. Parkinson’s at risk syndrome: can Parkinson’s disease be predicted? Mov Disord. 2010;25 Suppl 1:S89–S93.
- Siderowf A, Jennings D, Eberly S, et al. Impaired olfaction and other prodromal features in the Parkinson At-Risk Syndrome Study. Mov Disord. 2012;27(3):406–412.
- Jennings D, Siderowf A, Stern M, et al. Imaging prodromal Parkinson disease: the Parkinson Associated Risk Syndrome Study. Neurology. 2014;83(19):1739–1746.
- Chahine LM, Weintraub D, Hawkins KA, et al. Cognition in individuals at risk for Parkinson’s: Parkinson associated risk syndrome (PARS) study findings. Mov Disord. 2016;31(1):86–94.
- Jennings D, Siderowf A, Stern M, et al. Conversion to Parkinson Disease in the PARS Hyposmic and Dopamine Transporter-Deficit Prodromal Cohort. JAMA Neurol. 2017;74(8):933–940.
- Postuma RB, Arnulf I, Hogl B, et al. A single-question screen for rapid eye movement sleep behavior disorder: a multicenter validation study. Mov Disord. 2012;27(7):913–916.
- Postuma RB, Pelletier A, Berg D, et al. Screening for prodromal Parkinson’s disease in the general community: a sleep-based approach. Sleep Med. 2016;21:101–105.
- Romero K, Conrado D, Burton J, et al. Molecular neuroimaging of the dopamine transporter as a patient enrichment biomarker for clinical trials for early Parkinson’s disease. Clin Transl Sci. 2019;12(3):240–246.
- Vilas D, Iranzo A, Tolosa E, et al. Assessment of alpha-synuclein in submandibular glands of patients with idiopathic rapid-eye-movement sleep behaviour disorder: a case-control study. Lancet Neurol. 2016;15(7):708–718.
- Doppler K, Jentschke HM, Schulmeyer L, et al. Dermal phospho-alpha-synuclein deposits confirm REM sleep behaviour disorder as prodromal Parkinson’s disease. Acta Neuropathol. 2017;133(4):535–545.
- Antelmi E, Donadio V, Incensi A, et al. Skin nerve phosphorylated alpha-synuclein deposits in idiopathic REM sleep behavior disorder. Neurology. 2017;88(22):2128–2131.
- Berg D. Transcranial ultrasound as a risk marker for Parkinson’s disease. Mov Disord. 2009;24 Suppl 2:S677–S683.
- De Marzi R, Seppi K, Hogl B, et al. Loss of dorsolateral nigral hyperintensity on 3.0 tesla susceptibility-weighted imaging in idiopathic rapid eye movement sleep behavior disorder. Ann Neurol. 2016;79(6):1026–1030.
- Rolinski M, Griffanti L, Piccini P, et al. Basal ganglia dysfunction in idiopathic REM sleep behaviour disorder parallels that in early Parkinson’s disease. Brain. 2016;139(Pt 8):2224–2234.
- Mollenhauer B, Batrla R, El-Agnaf O, et al. A user’s guide for alpha-synuclein biomarker studies in biological fluids: Perianalytical considerations. Mov Disord. 2017;32(8):1117–1130.
- Sardi SP, Cedarbaum JM, Brundin P. Targeted therapies for Parkinson’s disease: from genetics to the clinic. Mov Disord. 2018;33(5):684–696.
- Lang AE, Espay AJ. Disease modification in Parkinson’s disease: current approaches, challenges, and future considerations. Mov Disord. 2018;33(5):660–677.
- Gallo P, Chuang-Stein C, Dragalin V, et al. Adaptive designs in clinical drug development—an Executive Summary of the PhRMA Working Group. J Biopharm Stat. 2006;16(3):275–283; discussion 85–91, 93–98, 311–312.
- Kairalla JA, Coffey CS, Thomann MA, Muller KE. Adaptive trial designs: a review of barriers and opportunities. Trials. 2012;13:145.
- Maetzler W, Liepelt I, Berg D. Progression of Parkinson’s disease in the clinical phase: potential markers. Lancet Neurol. 2009;8(12):1158–1171.
- DATATOP: a multicenter controlled clinical trial in early Parkinson’s disease. Parkinson Study Group. Arch Neurol. 1989;46(10):1052–1060.
- Cedarbaum JM. Elephants, Parkinson’s disease, and proof-of-concept clinical trials. Mov Disord. 2018;33(5):697–700.
- Romero K, Ito K, Rogers JA, et al. The future is now: model-based clinical trial design for Alzheimer’s disease. Clin Pharmacol Ther. 2015;97(3):210–214.
- Knopman DS, Siemers ER, Bain LJ, et al. National Institute on Aging—Alzheimer’s Association Research Framework lays the groundwork for deeper understanding of Alzheimer’s disease. Alzheimers Dement. 2018;14(2):261–262.
- Jack CR, Jr., Bennett DA, Blennow K, et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018;14(4):535–562.
- Messmer MF, Wilhelm EE, Shoulson I. I-SPY 2 breast cancer trial as a model for innovation in Alzheimer disease therapies. JAMA Neurol. 2017;74(9):1027–1028.
- Brundin P, Barker RA, Conn PJ, et al. Linked clinical trials–the development of new clinical learning studies in Parkinson’s disease using screening of multiple prospective new treatments. J Parkinsons Dis. 2013;3(3):231–239.
- Brundin P, Wyse RK. The Linked Clinical Trials Initiative (LCT) for Parkinson’s disease. Eur J Neurosci. 2019;49(3):307–315.