
by Joana Jesus-Ribeiro, PhD; Luís Miguel Pires, MS; Ilda Patrícia Ribeiro, PhD; Olinda Rebelo, MD; Ricardo Pereira, MD; Francisco Sales, MD; Isabel Santana, PhD; António Freire, PhD; and Joana Barbosa Melo, PhD
Dr. Jesus-Ribeiro is with Neurology Department, Centro Hospitalar de Leiria in Leiria, Portugal. Drs. Jesus-Ribeiro, Ribeiro, and Melo and Mr. Pires are with Coimbra Institute for Clinical and Biomedical Research (iCBR) and Center of Investigation on Environment, Genetics, and Oncobiology (CIMAGO), Faculty of Medicine, University of Coimbra in Coimbra, Portugal. Drs. Ribeiro and Melo and Mr. Pires are also with Laboratory of Cytogenetics and Genomics, Faculty of Medicine, University of Coimbra in Coimbra, Portugal. Dr. Rebelo is with Neuropathology Laboratory, Neurology Department, Centro Hospitalar e Universitário de Coimbra in Coimbra, Portugal. Dr. Pereira is with Neurosurgery Department, Centro Hospitalar e Universitário de Coimbra in Coimbra, Portugal. Dr. Sales is with Epilepsy and Sleep Monitoring Unit, Neurology Department, Centro Hospitalar e Universitário de Coimbra in Coimbra, Portugal. Dr. Santana is with Neurology Department, Centro Hospitalar e Universitário de Coimbra in Coimbra, Portugal. Drs. Santana and Freire are with Faculty of Medicine, University of Coimbra in Coimbra, Portugal. Dr. Freire is also with Neurology Department, Luz Hospital in Coimbra, Portugal.
Funding: Funding was provided by Clinical Research Fellowship in Epilepsy, awarded by Tecnifar; Scientific Fellowship of the Portuguese League against Epilepsy; and a research grant awarded by the Center for Research in Environment, Genetics and Oncobiology (CIMAGO), Faculty of Medicine, University of Coimbra. These funding sources were used to support laboratory techniques. The funding sources did not influence the preparation of the data or the manuscript.
Disclosures: The authors have no conflicts of interest relevant to the contents of this article.
Innov Clin Neurosci. 2023;20(10–12):35–39.
Abstract
Objective: The advent of next-generation sequencing (NGS) enabled the detection of low-level brain somatic variants in postsurgical tissue of focal cortical dysplasia (FCD). The genetic background of FCD Type I remains elusive, while the mammalian target of rapamycin (mTOR) pathway seems to have a relevant role in the pathogenesis of FCD Type II. Our goal was to uncover information on the molecular basis of FCD, performing whole genome sequencing (WGS) in postsurgical tissue to detect candidate brain-specific somatic variants, and evaluate their clinical significance.
Design: WGS was performed using paired peripheral venous blood and postsurgical pathological brain deoxyribonucleic acid (DNA) samples. Libraries were prepared using the Roche KAPA HyperPrep polymerase chain reaction (PCR) free library preparation kit. Paired‐end 150bp reads were generated on the Illumina NovaSeq platform. The FASTQ files were processed using the nf-core sarek pipeline (version 3.0) to call somatic variants, which were then annotated with ANNOVAR. A screening strategy was applied to obtain relevant variants.
Results: Two female patients with drug-resistant epilepsy due to FCD who underwent surgical treatment were included. Regarding neuropathological diagnosis, one patient had FCD Type Ia and the other had FCD Type IIa. Five somatic nonsynonymous single nucleotide variants (SNVs) were detected using WGS, three in FCD Ia tissue (WDR24 p.Trp259Gly; MICAL1 p.Lys1036Arg; and KATNB1 p.Leu566Ile) and two in FCD IIa tissue (MATN4 p.Phe91Val and ANKRD6 p.His386Gln). All variants were predicted to be potentially pathogenic by at least two different tools. However, they were classified as variants of uncertain significance (VUS) according to the American College of Medical Genetics and Genomics (ACMG) criteria.
Conclusion: Brain-specific somatic missense variants were identified by NGS in new candidate genes (WDR24, MICAL1, KATNB1, MATN4, and ANKRD6) using postsurgical FCD tissue, which may contribute to further understanding of the genetic background of FCD. All the reported genes were previously related to epilepsy and/or malformations of central nervous system (CNS) and cortical development. However, the pathogenicity assessment of these variants and, consequently, their impact on clinical practice still poses an important challenge.
Keywords: Focal cortical dysplasia, drug-resistant epilepsy, next-generation sequencing, somatic variant, missense variant
Focal cortical dysplasia (FCD) is a heterogeneous group of cortical development malformations highly associated with drug-resistant epilepsy, and it is a common etiology among patients who undergo epilepsy surgery.1 The International League Against Epilepsy (ILAE) classification grades focal cortical dysplasia into Type I (cortical dyslamination), Type II (dyslamination plus dysmorphic neurons and balloon cells), and Type III (associated with other epileptogenic lesions).2
In the last few years, next-generation sequencing (NGS) has allowed the identification of de novo brain-specific somatic variants with low variant allelic frequencies in postsurgical FCD tissue.3,4 For FCD Type II, most of the pathogenic variants affected single genes regulating the mammalian target of rapamycin (mTOR) signaling pathway.3,5 However, new candidate genes involved in other circuits, such as insulin and Ras pathways, have also been reported.6 In contrast, the genetic basis of FCD Type I remains elusive, and it is doubtful whether the mTOR signaling cascade is a relevant target, suggesting that FCD subtypes may represent different molecular entities.3,4
Our goal was to uncover information on the molecular basis of FCD, performing whole genome sequencing (WGS) on postsurgical tissue to detect candidate brain-specific somatic variants and analyze their relevance in clinical practice.
Design
Two patients with drug-resistant epilepsy due to FCD who underwent surgical treatment were included. The resected specimen’s processing was performed according to international recommendations,7 and a part of the tissue was freshly frozen at -80ºC to be posteriorly used in the study. The histopathological diagnosis was executed based on the ILAE consensus classifications proposed by ad hoc task forces of the ILAE diagnostic methods commission.8,9
Peripheral venous blood was collected in an ethylenediaminetetraacetic acid (EDTA) tube (5mL) during the routine preoperative tests, and the deoxyribonucleic acid (DNA) was subsequently extracted using the Agilent DNA kit (#200600, Agilent Technologies; Santa Clara, California, United States). DNA was extracted from fresh frozen pathological brain tissue using QIAamp DNA mini kit (50) (p/n 51304, Qiagen; Valencia, California, United States) and High Pure Polymerase Chain Reaction (PCR) Template Preparation Kit (Roche GmbH; Mannheim, Germany), according to the manufacturer’s instructions. DNA concentration and purity were quantified by ultraviolet (UV) spectrophotometric analysis using a Nanodrop 1000 Spectrophotometer (Thermo Fisher Scientific; Waltham, Massachusetts, United States) and fluorometric quantification using a Qubit 3.0 (Life Technologies; Carlsbad, California, United States).
WGS was performed using matched blood/postsurgical brain DNA samples. Libraries were prepared using the Roche KAPA HyperPrep (KAPABIOSYSTEMS, Cape Town, South Africa). PCR free library preparation kit. Paired‐end 150bp reads were generated on the Illumina (San Diego, California, United States) NovaSeq platform. The FASTQ files were then processed using the nf-core sarek pipeline (version 3.0) to call somatic variants. Somatic single nucleotide variants (SNVs) were annotated using ANNOVAR. To obtain potentially deleterious variants, a screening process following some criteria was applied. Variants located in deep introns, synonymous variants, and variants identified with minor allele frequency greater than 0.05 in population databases were discarded. We retained missense SNVs predicted to be potentially pathogenic by at least two of the following algorithms: Polymorphism Phenotyping v2 (PolyPhen-2), Sorting Intolerant From Tolerant (SIFT), and MutationTaster.
All procedures were conducted in accordance with the Declaration of Helsinki and approved by the local ethics committee. Informed written consent was obtained from both patients.
Results
The detected somatic variants are presented in Table 1.


Patient 1. A 22-year-old, right-handed female patient, born from a twin pregnancy with preterm birth and with mild intellectual disability disorder, was diagnosed with structural focal epilepsy at the age of 14 years. Most of her seizures would occur on a monthly basis. Neurologic examination was unremarkable. Her epilepsy did not respond to several trials with antiseizure medications, and she was proposed for presurgical evaluation.
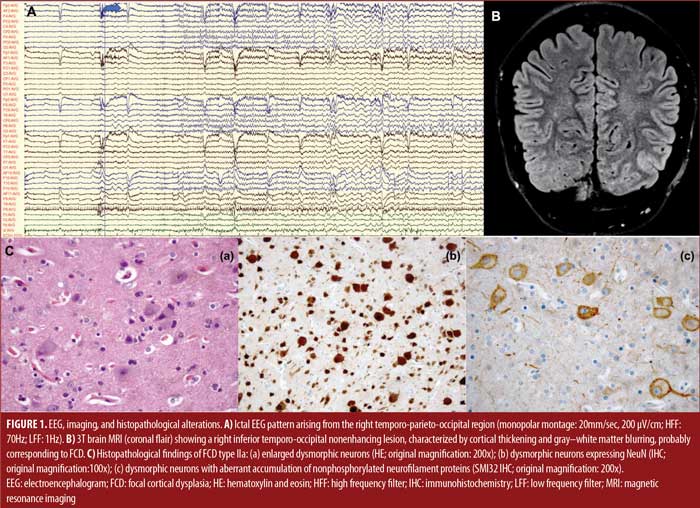
She was admitted to the Epilepsy Monitoring Unit, and she had five seizures arising from the right temporo-parieto-occipital region (Figure 1A), characterized by elementary visual aura (2 out of 5), followed by automotor seizure with impaired awareness, progressing to bilateral tonic-clonic seizure (1 out of 5). Brain 3T magnetic resonance imaging (MRI) showed a right inferior temporo-occipital focal cortical dysplasia (Figure 1B). The patient performed other standard complementary exams, namely ictal single-photon emission computed tomography (SPECT), interictal positron emission tomography (PET), and neuropsychological evaluation, which supported a lateralization to the right hemisphere without congruent localization. An invasive intracranial study with stereoelectroencephalography (SEEG) confirmed an extensive epileptogenic circuit, predominantly affecting the right temporo-occipital regions. SEEG-guided radiofrequency thermocoagulation was executed. However, a seizure relapse occurred after three months. Posteriorly, she underwent surgical resection of the dysplastic area plus right hippocampectomy. The neuropathological diagnosis pointed to FCD Type Ia. WGS identified three somatic missense variants classified as variants of uncertain significance (VUS) in the following genes: WD repeat domain 24 (WDR24) p.Trp259Gly; molecule interacting with CasL 1 (MICAL1) p.Lys1036Arg; and katanin regulatory subunit B1 (KATNB1) p.Leu566Ile (Table 1). Currently, the patient is Class III (worthwhile improvement), according to the Engel Epilepsy Surgery Outcome Scale.
Patient 2. A 41-year-old, right-handed female patient with no relevant clinical background except a family history of epilepsy (father and sister) was diagnosed with structural focal epilepsy at the age of three years. The patient had daily seizures, mainly during sleep, despite antiseizure medication. Neurologic examination was unremarkable.
She was admitted to the Epilepsy Monitoring Unit for presurgical evaluation. Video electroencephalogram (EEG) revealed nonmotor onset seizures, with behavior arrest and impaired awareness, occasionally evolving to a bilateral tonic-clonic seizure associated with a seizure pattern arising from the left frontal region with propagation to the left anterior temporal lobe. The first brain 1.5T MRIs were normal. Ictal SPECT showed a left frontal hyperperfusion, and interictal PET revealed a mild left frontal hypometabolism. Invasive EEG monitoring with subdural grids was also performed to complement the previous data.
The patient underwent epilepsy surgery twice. At first, a lesionectomy involving the left middle frontal region was executed, but 17 years later, the surgical resection was extended due to an increase in seizure frequency. The neuropathological diagnosis demonstrated FCD Type IIa (Figure 1C). WGS performed in the pathologic brain tissue from the second surgery identified two somatic missense variants classified as VUS in the genes matrilin 4 (MATN4) p.Phe91Val and ankyrin repeat domain 6 (ANKRD6) p.His386Gln (Table 1). Short-term postsurgical Engel classification was Class III, but the patient developed neuropsychiatric and behavioral alterations.
Discussion
We report the occurrence of rare somatic missense variants in human pathological brain tissue from two patients with focal drug-resistant epilepsy due to FCD. FCD may result from somatic gene mutation(s) in a single or small subset of neuroglial progenitor cells in the telencephalic ventricular zone during embryogenesis, forming an abnormal dysplastic area surrounded by a normal cortex.10,11 Phenotype variability depends on the embryonic stage in which the postzygotic mutational event occurred, and consequently on the fraction of brain cells carrying it.10,11 A few studies showed that somatic variants in different genes can co-occur in one patient and accumulate during the embryonic cell proliferation.4,6
The genetic background of FCD Type I is still a field of uncertainty since the reported data encompass brain mosaic variants targeting a pleomorphic spectrum of genes.4 We found three nonsynonymous SNVs in our FCD Ia tissue, involving the genes WDR24, MICAL1, and KATNB1 (Table 1).
The gene WDR24 belongs to GATOR2, a pentameric protein complex thought to inhibit the GATOR1 complex.12 The GATOR1 complex is an upstream regulator of the mTOR Complex 1 (mTORC1) pathway, which is involved in essential cellular functions, including protein synthesis, cell growth, cell migration, and cell proliferation.12 Mutations in the GATOR1 complex genes (DEPDC5, NPRL2, and NPRL3) lead to mTORC1 hyperactivation. However, to date, no pathogenic gain-of-function mutations have been found in GATOR2 complex genes.12 Weckhuysen et al12 reported two germline missense variants in WDR24 (p.Pro573Leu, p.Arg241His) in familial focal epilepsy. Although they were predicted to be disease causing and possibly/probably damaging by MutationTaster and PolyPhen-2, respectively, there was insufficient evidence to support their pathogenicity, and they were classified as VUS.12 In a broad array of Drosophila tissues, Wdr24 proved to be a critical effector of the GATOR2 complex that promotes TORC1 activity and cellular growth.13
Disease-causing variants in the MICAL1 gene have previously been reported in autosomal dominant lateral temporal epilepsy (p.Gly150Ser, p.Ala1065fs), which significantly increased MICAL1 oxidoreductase activity and induced cell contraction in cell-based assays.14 MICAL1 oxidoreductase activity induces the disassembly of actin filaments, thereby regulating the organization of the actin cytoskeleton in developing and adult neurons.14 This suggests that dysregulation of the actin cytoskeleton dynamics is a likely mechanism by which MICAL1 variants lead to focal epilepsy.14
KATNB1 encodes the regulatory subunit (p80) of the microtubule-severing enzyme katanin, and it has a fundamental role in human cerebral cortical development and pathology.15,16 An exome sequencing analysis of over 2,000 children with complex malformations of cortical development identified homozygous deleterious mutations in KATNB1.15 Knockdown of KATNB1 orthologs in zebrafish (katnb1) and Drosophila (kat80) recapitulated the human phenotype.15 In Drosophila, kat80 is essential for the formation of the mitotic spindle, and its loss results in supernumerary centrosomes and delayed anaphase onset, preferentially affecting asymmetrically dividing neuroblasts in vivo.15 Furthermore, kat80 depletion results in neuronal dendritic arborization defects, affecting neural architecture.15
Somatic gain-of-function variants in MTOR and its activators and germline, somatic, and second-hit mosaic loss-of-function variants in its related repressors have been reported as the major causes in FCD Type II.4 However, other potentially deleterious somatic variants in novel genes (IRS1, RAB6B, ZNF337, RALA, HTR6) have also been identified,6 and negative sequencing results are frequent,4 bringing to light the complexity of FCD Type II pathogenesis. In our FCD Type IIa tissue, we identified two nonsynonymous SNVs in the genes MATN4 and ANKRD6.
MATN4 belongs to a family of noncollagenous extracellular matrix proteins with a modular composition consisting of von Willebrand factor A (vWFA)-like domains, epidermal growth factor (EGF)-like domains, and an α-helical coiled-coil oligomerization domain. Mouse matrilin 4 gene expression was high in the brain.17 The missense variant MATN4 p.Gly172Ala has been related to a structural malformation of the human forebrain (holoprosencephaly).18 The remaining phenotype also included global developmental delay, microcephaly, and epilepsy, among others.18
Diversin, encoded by Ankrd6, is an ankyrin repeat protein that activates the noncanonical Wnt/planar cell polarity (PCP) pathway and simultaneously inhibits the canonical Wnt/β-catenin pathway.19 Rare missense mutations in ANKRD6 (p.Pro548Leu, p.Arg632His) could affect a balanced reciprocal antagonism between both Wnt pathways in neurulation and act as predisposing factors to neural tube defects.19 Also, Ankrd6 is expressed prominently in the mouse-developing brain.20 In embryos, expression is maximal in ventricular zones of neuronal proliferation and intermediate zones of neuronal migration and extends to postmigratory neuronal fields during the postnatal period.20
Limitations. The implementation of NGS created an ever-increasing catalogue of genetic findings that were accompanied by new challenges in sequence interpretation, particularly for missense variants.21 Although our variants were predicted to be potentially pathogenic by at least two algorithms, they were classified as VUS according to the ACMG criteria,21 making it impossible to confirm the role of these variants in FCD development without functional experiments. Sequencing at high coverage may lead to a substantial amount of artifactual, low-level somatic variants, which may significantly lower positive predictive values.4 Another limitation of our study is the small sample size. Thus, further research is needed to complement the genomic background of FCD.
Conclusion
The analysis of postsurgical tissue by NGS may contribute to identifying somatic variants in new candidate genes to further understand the genetic basis of FCD. The pathogenicity assessment of these variants and, consequently, their impact on clinical practice remains a challenge.
References
- Blumcke I, Spreafico R, Haaker G, et al. Histopathological findings in brain tissue obtained during epilepsy surgery. N Engl J Med. 2017;377(17):1648–1656.
- Blümcke I, Spreafico R. An international consensus classification for focal cortical dysplasias. Lancet Neurol. 2011;10(1):26–27.
- Baldassari S, Ribierre T, Marsan E, et al. Dissecting the genetic basis of focal cortical dysplasia: a large cohort study. Acta Neuropathol. 2019;138(6):885–900.
- Jesus-Ribeiro J, Pires LM, Melo JD, et al. Genomic and epigenetic advances in focal cortical dysplasia types I and II: a scoping review. Front Neurosci. 2020;14:580357.
- Lim JS, Kim WI, Kang HC, et al. Brain somatic mutations in MTOR cause focal cortical dysplasia Type II leading to intractable epilepsy. Nat Med. 2015;21(4):395–400.
- Zhang Z, Gao K, Liu Q, et al. Somatic variants in new candidate genes identified in focal cortical dysplasia Type II. Epilepsia. 2020;61(4):667–678.
- Blümcke I, Aronica E, Miyata H, et al. International recommendation for a comprehensive neuropathologic workup of epilepsy surgery brain tissue: a consensus Task Force report from the ILAE Commission on Diagnostic Methods. Epilepsia. 2016;57(3):348–358.
- Blümcke I, Thom M, Aronica E, et al. The clinicopathologic spectrum of focal cortical dysplasias: a consensus classification proposed by an ad hoc Task Force of the ILAE Diagnostic Methods Commission. Epilepsia. 2011;52(1):158–174.
- Najm I, Lal D, Alonso Vanegas M, et al. The ILAE consensus classification of focal cortical dysplasia: an update proposed by an ad hoc task force of the ILAE diagnostic methods commission. Epilepsia. 2022;63(8):1899–1919.
- Iffland PH 2nd, Crino PB. Focal cortical dysplasia: gene mutations, cell signaling, and therapeutic implications. Annu Rev Pathol. 2017;12:547–571.
- D’Gama AM, Woodworth MB, Hossain AA, et al. Somatic mutations activating the mTOR pathway in dorsal telencephalic progenitors cause a continuum of cortical dysplasias. Cell Rep. 2017;21(13):3754–3766.
- Weckhuysen S, Marsan E, Lambrecq V, et al. Involvement of GATOR complex genes in familial focal epilepsies and focal cortical dysplasia. Epilepsia. 2016;57(6):994–1003.
- Cai W, Wei Y, Jarnik M, et al. The GATOR2 component Wdr24 regulates TORC1 activity and lysosome function. PLoS Genet. 2016;12(5):e1006036.
- Dazzo E, Rehberg K, Michelucci R, et al. Mutations in MICAL-1 cause autosomal-dominant lateral temporal epilepsy. Ann Neurol. 2018;83(3):483–493.
- Mishra-Gorur K, Çağlayan AO, Schaffer AE, et al. Mutations in KATNB1 cause complex cerebral malformations by disrupting asymmetrically dividing neural progenitors. Neuron. 2014;84(6):1226–1239.
- Hu WF, Pomp O, Ben-Omran T, et al. Katanin p80 regulates human cortical development by limiting centriole and cilia number. Neuron. 2014;84(6):1240–1257.
- Wagener R, Kobbe B, Aszódi A, et al. Characterization of the mouse matrilin-4 gene: a 5’ antiparallel overlap with the gene encoding the transcription factor RBP-l. Genomics. 2001;76(1–3):89–98.
- Alazami AM, Patel N, Shamseldin HE, et al. Accelerating novel candidate gene discovery in neurogenetic disorders via whole-exome sequencing of prescreened multiplex consanguineous families. Cell Rep. 2015;10(2):148–161.
- Allache R, Wang M, De Marco P, et al. Genetic studies of ANKRD6 as a molecular switch between Wnt signaling pathways in human neural tube defects. Birth Defects Res A Clin Mol Teratol. 2015;103(1):20–26.
- Tissir F, Bar I, Goffinet AM, et al. Expression of the ankyrin repeat domain 6 gene (Ankrd6) during mouse brain development. Dev Dyn. 2002;224(4):465–469.
- Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424.