
 by Thomas Shiovitz, MD; William M. Greenberg, MD; Changzheng Chen, PhD; Giovanna Forero, MA; and Carl P. Gommoll, MS
by Thomas Shiovitz, MD; William M. Greenberg, MD; Changzheng Chen, PhD; Giovanna Forero, MA; and Carl P. Gommoll, MS
Dr. Shiovitz with the California Neuroscience Research Medical Group Inc, Sherman Oaks, California; and Dr. Greenberg, Mr. Chen, Ms. Forero, and Mr. Gommoll are from Forest Research Institute, Jersey City, New Jersey.
Innov Clin Neurosci. 2014;11(1–2):10–22
Funding: This research was funded by Forest Laboratories, Inc. and Pierre Fabre Médicament.
Clinicaltrials.gov registry: NCT01085812
Financial disclosures: Dr. Shiovitz is a consultant for Eli Lilly and has received financial compensation from Forest Research Laboratories, Inc. for conducting this study. William M. Greenberg, Changzheng Chen, Giovanna Forero, and Carl P. Gommoll are employees of Forest Research Institute.
Key words: Levomilnacipran, antidepressants, serotonin and norepinephrine reuptake inhibitor (SNRI), major depressive disorder, major depressive disorder relapse
Abstract
Objective: Major depressive disorder is often chronic, with relapse and recurrence common. Levomilnacipran extended-release is a potent and selective serotonin and reuptake inhibitor approved in the United States for treatment of major depressive disorder in adults. The objective of this study (NCT01085812) was to evaluate the efficacy, safety, and tolerability of levomilnacipran extended-release in the prevention of relapse in patients with major depressive disorder.
Design: A 24-week Phase III randomized, double-blind, controlled trial comparing levomilnacipran extended-release 40–120mg/day with placebo for relapse prevention in patients with major depressive disorder who had responded to 12-week, open-label treatment with levomilnacipran extended-release. Statistical power was calculated on the assumption that 38 percent of placebo and 20 percent of levomilnacipran extended-release patients would relapse.
Setting: Thirty-six outpatient study centers throughout the United States and Canada.
Participants: Of 348 patients who met randomization criteria and entered double-blind treatment, three discontinued prior to treatment, 112 were randomized to placebo, and 233 to levomilnacipran extended-release.
Measurements: Primary outcome: Time to relapse was analyzed using the Cox proportional hazard-regression model with treatment group and baseline Montgomery-Åsberg Depression Rating Scale score as explanatory variables. Safety was also evaluated.
Results: Time to relapse was longer for levomilnacipran extended-release versus placebo (hazard ratio [95% confidence interval] = 0.68 [0.40, 1.17]), but the treatment difference was not statistically significant (P=0.165). A relatively low percentage of patients from either group relapsed (placebo=20.5%, levomilnacipran extended-release=13.9%).
Conclusion: This study did not detect between-treatment group differences, potentially due to lower than expected relapse rates in the placebo group. Levomilnacipran extended-release was generally well tolerated.
Background
Major depressive disorder (MDD) is often a chronic condition, characterized by a relapsing and recurrent course of illness. In a large trial designed to assess the efficacy of sequential acute treatments for MDD, the six-month relapse rate was between 34 and 83 percent.[1] The risk of additional episodes increases with each new episode; two major depressive episodes are associated with a 70-percent risk of a third, while three episodes are associated with a 90-percent risk of a fourth.[2] Relapse of depression, defined as the reemergence of symptoms during recovery from an index episode of depression,[3] is associated with several risk factors, including the presence of residual depressive symptoms following treatment, a history of multiple prior depressive episodes, and a diagnosis of recurrent depression.[4]
In patients with recurrent depression, maintenance treatment with antidepressant pharmacotherapy is recommended to prevent depressive relapse.[5] Results from randomized, placebo-controlled, double-blind trials of antidepressants have demonstrated significantly lower rates of relapse and/or a longer time to relapse with active agents compared with placebo.[6–8]
Current treatment guidelines advise that some patients remain on treatment with the same drug at the same dose level that effectively treated the index episode of depression for at least 4 to 9 months to prevent relapse; additionally, for patients who have had three or more prior major depressive episodes, full-dose maintenance treatment for an indeterminate duration may be recommended.[5]
Levomilnacipran (1S, 2R-milnacipran) is a potent and selective serotonin and norepinephrine reuptake inhibitor (SNRI) that was recently approved in the United States for treatment of MDD in adults; an extended-release (ER) formulation was developed to allow for once-daily dosing. The recommended dose of levomilnacipran ER is 40 to 120mg/day.[9] Levomilnacipran has greater potency for inhibiting the norepinephrine transporter relative to the serotonin transporter;10 conversely, the SNRIs duloxetine,[10] venlafaxine,[10] and desvenlafaxine[11] show greater preference for serotonin reuptake inhibition. The efficacy and safety of levomilnacipran ER in the treatment of MDD have been evaluated in five Phase II/III studies, four of which met the prespecified primary efficacy endpoint,[12–15] change from baseline in the Montgomery-Åsberg Depression Rating Scale (MADRS).[16] In the other study, conducted early in the development program, levomilnacipran ER showed greater numerical improvement versus placebo on the primary efficacy endpoint, but the difference between groups was not statistically significant.[17] Additionally, the long-term safety and tolerability of levomilnacipran ER was evaluated in a 48-week open-label study.[18] Levomilnacipran ER, at the doses evaluated in these trials, has been shown to be generally safe and well tolerated. The objective of the current study (www.ClinicalTrials.gov, NCT01085812) was to evaluate the efficacy, safety, and tolerability of levomilnacipran ER in the prevention of relapse in patients with MDD.
Patients and Methods
The study was conducted at 36 centers in the United States and Canada between March 2010 and October 2011 in full compliance with United States Food and Drug Administration guidelines for Good Clinical Practice and the Declaration of Helsinki. The appropriate institutional review board at each study center approved the protocol and all patients provided written informed consent.
Study design. This was a Phase III, multicenter, randomized, double-blind, placebo-controlled study in adult outpatients with MDD. The study was up to 39 weeks in duration and consisted of a one-week no-drug screening period, 12-week open-label treatment period with flexible-dose levomilnacipran ER 40 to 120mg/day, 24-week double-blind treatment period with fixed-dose levomilnacipran ER (40, 80, or 120mg/day) or placebo, and a two-week double-blind down-taper period (Figure 1). Patients who completed open-label treatment and met the prespecified response criteria were eligible to enter double-blind treatment with levomilnacipran ER or placebo. Patients randomized to levomilnacipran ER continued at the same dosage (40, 80, or 120mg/day) that they were receiving at the end of the open-label treatment period; patients randomized to placebo were gradually down-tapered during the first week after randomization and received placebo thereafter. Patients who did not meet randomization criteria or who prematurely discontinued from open-label or the double-blind treatment were eligible to enter a two-week down-taper treatment period if medically appropriate.

In open-label treatment, levomilnacipran ER was initiated at 20mg/day for Days 1 and 2, and increased to 40mg/day for Days 3 to 7. At the end of Week 1, 2, or 4, dose could be increased to 80mg/day; for patients increased to 80mg/day, dose could be further increased to 120mg/day at Week 2 or 4 based on the Investigator’s judgment of a patient’s response and tolerability. If significant tolerability issues arose following an increase, the dose could be decreased in 40-mg decrements at any time during open-label treatment; the minimum allowable dose was 40mg/day. No dose increases were allowed after Week 4 of open-label treatment.
Patients who met criteria for response after the 12-week open-label treatment period were randomized (2:1) to 24 weeks of double-blind treatment with fixed-dose levomilnacipran ER or placebo. Responders were defined as patients who met the following criteria at both open-label Week 10 and the randomization visit (Week 12): MADRS total score of 12 or less and Clinical Global Impressions-Improvement (CGI-I)[19] score of 2 or less.
Patients were randomized using a computer-generated list of numbers and assigned to identically appearing treatment. Study staff and patients were blinded to allocation of the investigational product throughout double-blind treatment and down-taper periods. The blind was maintained via a secured randomization code list and was broken only in case of emergency; unblinding disqualified a patient from further study participation.
Key inclusion criteria. Male and female outpatients 18 to 65 years of age were eligible to participate in the study if they met the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision (DSM-IV-TR) criteria for MDD with an ongoing depressive episode of at least four weeks’ duration; the diagnosis was confirmed by the Mini International Neuropsychiatric Interview (MINI)[20] and a MADRS total score of 22 or greater. Patients were required to have normal physical examination results, clinical laboratory determinations, and electrocardiogram (ECG) findings, or abnormal results that were judged by the Investigator to be not clinically significant.
Key exclusion criteria. Patients were excluded if they had been diagnosed with any DSM-IV-TR Axis I disorder other than MDD within six months of screening (comorbid generalized anxiety disorder, social anxiety disorder, and/or specific phobias were permitted), had a history of various psychiatric disorders (e.g., mania, depressive episode with psychotic features, obsessive-compulsive disorder, schizophrenia, borderline or antisocial personality disorder, cognitive disorder), or had substance abuse or dependence within six months of screening. Additional reasons for exclusion included the following: history of nonresponse to two or more adequate treatment trials with antidepressants; certain medical conditions (e.g., cardiovascular, pulmonary, hepatic, gastrointestinal, renal, endocrine, neurological, autoimmune, or infectious disease) that may have interfered with the conduct of the study or confounded interpretation of study results; history or ECG evidence of QTc prolongation or pulse rates or blood pressures outside of normal limits at screening; female patients of child-bearing potential who were pregnant, breastfeeding, or not currently using a medically acceptable method of contraception; and significant risk of suicide determined by the Investigator or information generated from the Columbia-Suicide Severity Rating Scale (C-SSRS),21 suicide attempt within the past year, or a score of 5 or greater on MADRS Item 10 (Suicidal Thoughts).
Assessments. The prospectively defined primary efficacy parameter was time to relapse during the double-blind treatment period measured in days from the date of randomization. Relapse was defined as any of the following criteria: 1) MADRS total score of 22 or greater at two consecutive visits; 2) increase of 2 or greater points in the CGI-I score relative to the double-blind baseline score at two consecutive visits; 3) discontinuation from the study due to insufficient therapeutic response; or 4) MADRS Item 10 score of 4 or greater. The date of the first assessment was defined as the relapse date in cases where relapse was based on MADRS or CGI-I scores from two consecutive visits.
Additional efficacy parameters included MADRS total score, the Clinical Global Impressions-Severity (CGI-S),[19] CGI-I, and the Sheehan Disability Scale (SDS).[22] During the open-label period, assessments were conducted at baseline, Week 1, Week 2, and every two weeks thereafter, with the exception of the SDS, which was conducted at baseline and every four weeks thereafter. During the 24-week double-blind treatment period, assessments were conducted at baseline, after Weeks 1 and 2, every two weeks for 10 weeks, and then every four weeks for the remaining 12 weeks of treatment; the SDS was conducted at baseline and every four weeks of double-blind treatment.
Safety assessments included recording of spontaneously reported or observed adverse events (AEs), physical examinations, vital signs, clinical laboratory evaluations, ECGs and the C-SSRS (evaluated at each visit). AEs were classified by preferred term; determination of severity and causality was made by the Investigator. An AE occurring during the open-label treatment period was considered a treatment-emergent AE (TEAE) if it was not present before the first open-label dose of levomilnacipran ER or was present but increased in severity during open-label treatment. An AE occurring during the double-blind treatment period was considered a TEAE if it was not present before the first dose of open-label levomilnacipran ER or was present but increased in severity during double-blind treatment. An AE occurring during the double-blind treatment period was considered a newly emergent AE (NEAE) if it was not present before the first dose of double-blind study drug or was present but increased in severity during the double-blind treatment period.
Statistical analysis. Projected enrollment to determine the number of patients needed to provide adequate statistical power to conduct the analysis was based on the assumption that 38 percent of placebo and 20 percent of levomilnacipran ER patients would relapse and the responder rate would be 52 percent during the open-label period. Based on these assumptions, it was expected that approximately 700 patients should be enrolled in open-label treatment and that approximately 360 patients would be randomized to double-blind treatment to provide adequate power (90% power with an alpha level of 0.05 for time to relapse) for analysis.
Populations in the open-label period consisted of the Safety Population (all patients who enrolled in the study and received at least 1 dose of open-label levomilnacipran ER) and the Intent-to Treat (ITT) Population (all open-label Safety Population who also had at least 1 MADRS total score assessment during open-label treatment). Populations in the double-blind period consisted of the Safety Population (all patients who completed open-label treatment, were randomized to double-blind treatment, and received at least 1 dose of double-blind study drug) and the ITT Population (all patients in the double-blind safety population who also had at least 1 MADRS total score assessment after randomization). Safety analyses were performed using the relevant Open-Label or Double-Blind Safety Population; efficacy analyses were based on the relevant ITT Population. The open-label treatment period started with the first dose of open-label levomilnacipran ER and ended with early termination or last assessment; the double-blind period started with the first dose of double-blind study drug and ended with early termination or last assessment.
Demographic parameters and other baseline characteristics were summarized overall for the Open-Label Safety Population and by treatment group for the Double-Blind Safety and Double-Blind ITT Populations. Double-blind treatment group comparisons were made using an analysis of variance (ANOVA) model with treatment group and study center as factors for continuous variables and the Cochran-Mantel-Haenszel test, controlling for study center, for categorical variables.
Time to relapse, the primary efficacy parameter, was defined as the number of days from the randomization date to when a patient met any relapse criterion during the double-blind treatment period; patients who relapsed were considered study completers. The primary analysis was carried out using the Cox proportional hazard-regression model with treatment group and baseline MADRS score as explanatory variables. Patients who did not meet relapse criteria were censored at the time of completion or discontinuation from the study. Kaplan-Meier estimates and curves for cumulative rate of relapse were generated for the double-blind treatment period.
The baseline for additional efficacy parameters in the double-blind treatment period was defined as the last nonmissing efficacy assessment before randomization. Descriptive statistics were provided by visit for additional efficacy parameters in the Open-Label and Double-Blind ITT populations using the last observation carried forward (LOCF) and observed cases (OC) approaches; the last CGI-S assessment before randomization was used as the baseline for CGI-I analysis. Additional efficacy parameters were analyzed based on all postbaseline scores using the OC approach without imputation of missing values. All statistical tests were two-sided hypothesis tests performed at the five-percent level of significance; all confidence intervals (CIs) were two-sided 95-percent CIs.
Results
A total of 1,066 patients were screened for eligibility; the number of patients who enrolled in the study and received 1 or more doses of levomilnacipran ER (n=734) exceeded the number projected to provide adequate statistical power (~700 patients); however, only 348 patients met randomization criteria and entered double-blind treatment, which was less than the 360 patients projected to adequately power the study. Three patients discontinued the study before receiving double-blind treatment; there were 345 patients in the Double-Blind Safety Population and 342 patients in the Double-Blind ITT Population (Table 1).

The most common reasons for early discontinuation of patients in the open-label treatment period were AEs and withdrawal of consent (Table 2); the most common reasons for early discontinuation in the double-blind treatment period were withdrawal of consent and lost to follow-up (Table 2). Baseline demographic characteristics and history of MDD were similar for the open-label and double-blind treatment populations (Table 3); there were no imbalances between placebo and levomilnacipran ER groups at baseline of double-blind treatment.

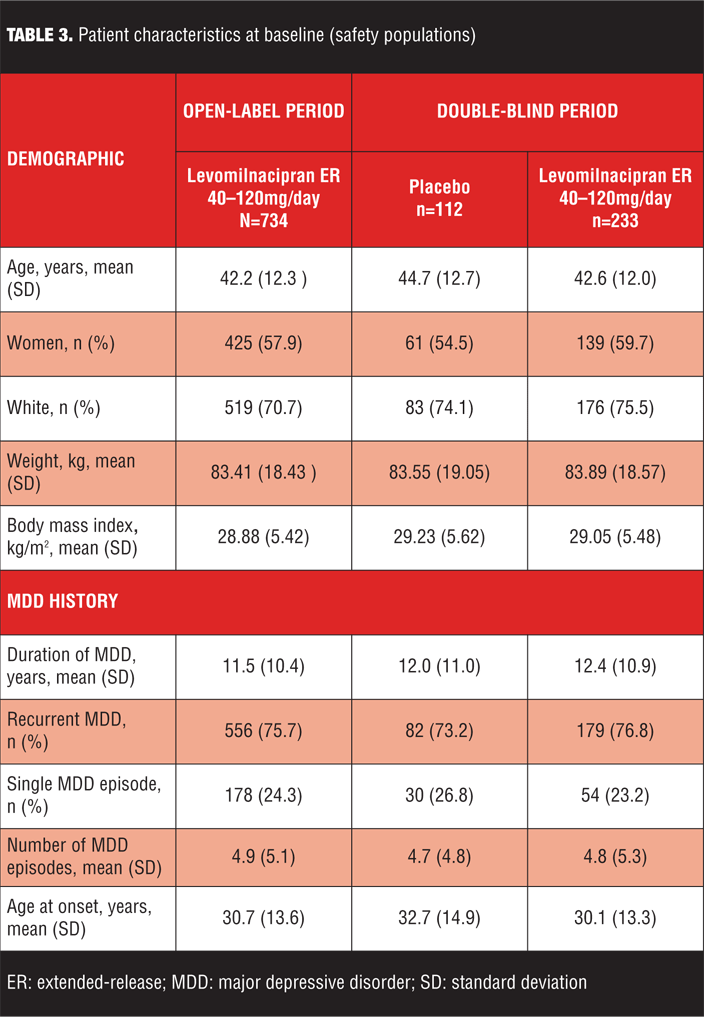
Efficacy assessments. Symptomatic improvement. No definite efficacy conclusions can be made for the open-label treatment period due to the lack of a placebo group for comparison; however, considerable symptomatic improvement appeared to occur as demonstrated by change from baseline on efficacy measures (Table 4). Low double-blind baseline scores indicated that the Double-Blind ITT population was relatively asymptomatic following open-label treatment with levomilnacipran ER (Table 4). Approximately 87 percent of patients were in remission (defined as MADRS total score of 10 or less) at the beginning of double-blind treatment, and there were no significant differences in mean score change between treatment groups on any efficacy measure (Table 4). During double-blind treatment, patients maintained the improvement observed at the end of open-label treatment and a much lower percentage of patients relapsed (placebo=20.5%, levomilnacipran ER=13.9%) than the percentage projected for sample size calculations (projected relapse: placebo=38%, levomilnacipran ER=20%).

Primary efficacy: time to relapse. Time to relapse was longer in the levomilnacipran ER group than in the placebo group (hazard ratio [95% CI]=0.68 [0.40, 1.17]), but the difference was not statistically significant (P=0.165) (Figure 2). Kaplan-Meier curves showed an initial numerical separation between treatment groups 30 days after the initiation of double-blind treatment, with an increased rate of relapse in the placebo group relative to levomilnacipran ER. Further numerical increase in the rate of relapse was observed for the placebo group relative to levomilnacipran ER at Day 140 and beyond, but rates of relapse were relatively low in both treatment groups.
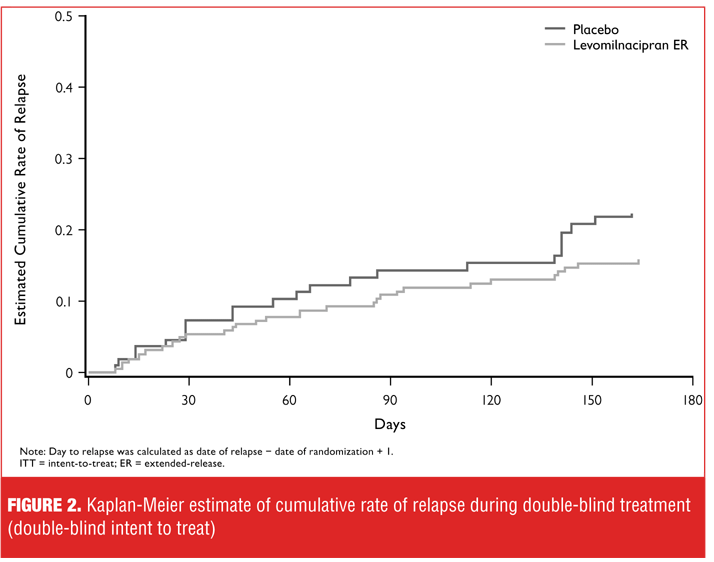
A series of post-hoc analyses were performed to identify any patient subgroups that might show greater between-treatment group differences. Increasing severity of depression, as measured by MADRS total score at study entry, was associated with progressively greater reduction in risk of relapse for levomilnacipran ER-treated patients relative to placebo (Table 5). In patients with the most severe depression (MADRS score of 36 or more), treatment with levomilnacipran ER was associated with significantly lower risk of relapse relative to placebo. Similarly, greater residual symptomatology (MADRS total score of 4 or more) following open-label treatment or a longer history of MDD also appeared to be associated with greater risk reduction for relapse in patients treated with levomilnacipran ER compared with placebo (data not shown).
Safety assessments. The mean open-label treatment duration was 69 days; the mean double-blind treatment duration was 136 days for placebo and 130 days for levomilnacipran ER. During open-label treatment, the overall mean daily dose of levomilnacipran ER was 79mg/day; 47 percent of patients received 120mg as the final daily dose. The overall mean final daily dose was 90mg.
Treatment-emergent adverse events. An overall summary of AEs reported during open-label and double-blind treatment is presented in Table 6. During open-label treatment, 10.9 percent (80/734) of the subjects discontinued due to AEs; most TEAEs were of mild or moderate intensity and 72 percent were considered by the Investigator to be related to levomilnacipran ER. During double-blind treatment, most TEAEs and NEAEs were of mild or moderate intensity; all severe TEAEs reported in both treatment groups were also NEAEs (placebo=9.8%; levomilnacipran ER=6.4%).
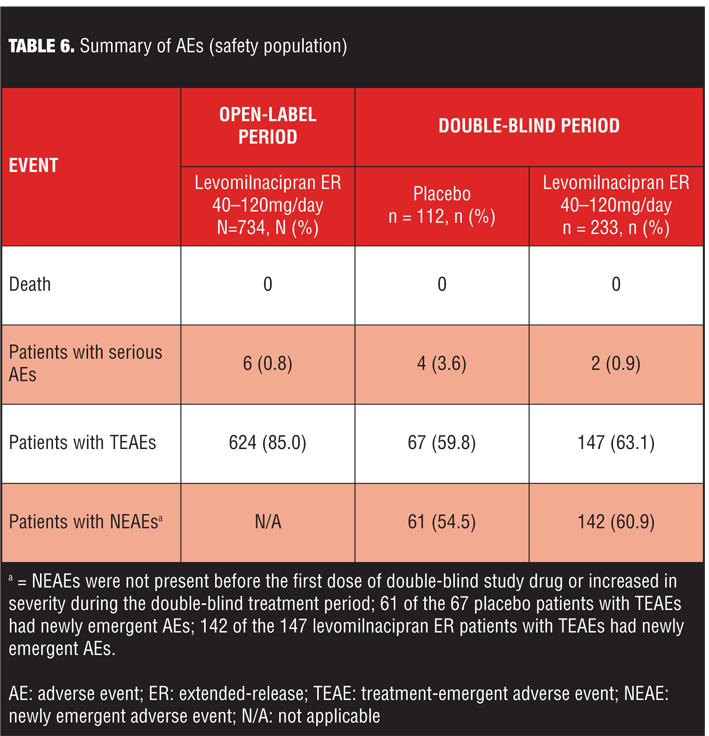
TEAEs were considered related to study drug in 25 percent of placebo patients and 28 percent of levomilnacipran ER patients during double-blind treatment.
Common TEAEs (5% or more of any treatment group) are shown in Table 7. During open-label treatment, TEAEs that led to the discontinuation of 3 or more patients included nausea (11); headache (7); urinary hesitation (5); dysuria, heart rate increased, and rash (4 patients each); and constipation, ejaculation disorder, hypertension, insomnia, and urinary retention (3 patients each). During double-blind treatment, three placebo patients and eight levomilnacipran ER patients prematurely discontinued because of TEAEs; no TEAE leading to discontinuation was reported by more than one patient in either treatment group.
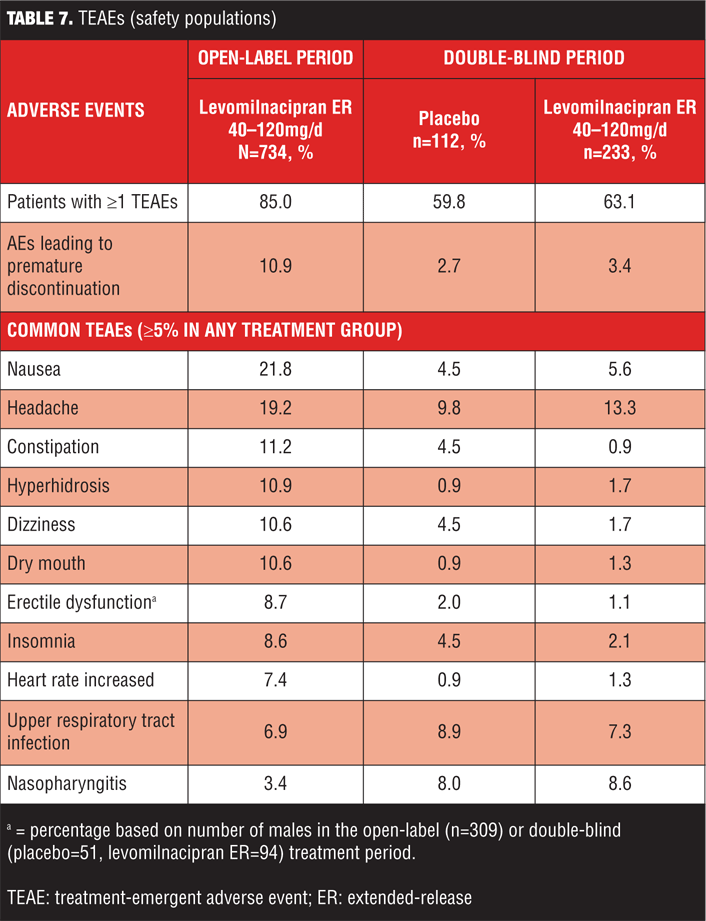
During open-label treatment, serious AEs (SAEs) were reported by six patients: hypertension; chest pain/troponin I increased/blood creatine phosphokinase MB increased/drug abuse; suicidal behavior; intentional self-injury/suicide attempt; cholelithiasis/acute pancreatitis; and ankle fracture/fall were reported in one patient each. The SAEs of chest pain, troponin I increased, and blood creatine phosphokinase MB increased that occurred in one patient was considered related to the concurrent SAE of drug abuse that led to premature discontinuation from the study. Only 1 SAE (hypertension) was considered to be related to levomilnacipran ER. This patient and three others with nonrelated SAEs (concurrent drug abuse, suicidal behavior; intentional self-injury/suicide attempt in 1 patient each) prematurely discontinued from the study.
During double-blind treatment, SAEs were reported by four placebo patients (cellulitis; blood pressure increased; pneumonia; drug sensitivity/deep vein thrombosis in 1 patient each) and two levomilnacipran ER patients (noncardiac chest pain and intestinal ischemia in 1 patient each). Noncardiac chest pain in one levomilnacipran ER patient was the only SAE considered related to study drug, but it did not result in premature discontinuation; two placebo patients prematurely discontinued the study due to SAEs.
Vital signs, clinical laboratory values, and electrocardiography. Mean changes in vital signs, clinical laboratory values, and ECGs were not clinically meaningful; changes were similar during the open-label and double-blind treatment periods, and they were similar between the placebo and levomilnacipran ER groups during double-blind treatment. No clinically meaningful changes in liver enzyme parameters were observed in patients during the open-label or double-blind treatment. Slight mean increases in alanine aminotransferase (ALT), aspartate aminotransferase (AST), and alkaline phosphatase were observed during open-label and double-blind treatment in the levomilnacipran ER group. Across both treatment periods, no patient met the criteria for Hy’s Law (ALT or AST of 3 or more × upper limit of normal [ULN] and total bilirubin >2 × ULN in the absence of alkaline phosphatase >2 × ULN)[23]; no ALT and AST increases were SAEs, and none resulted in discontinuation from the study. Levomilnacipran ER was associated with a slight mean decrease in weight (~0.5kg in each treatment period) while placebo was associated with a slight mean 0.5kg increase during double-blind treatment, supporting the weight neutrality of levomilnacipran ER treatment.
Potentially clinically significant changes from the respective open-label or double-blind treatment baselines were low (<1%) for pulse rate or blood pressure in levomilnacipran ER-treated patients. During double-blind treatment, no patient in either treatment group had a postbaseline QTcB or QTcF interval >500 msec. Levomilnacipran ER- versus placebo-treated patients had a greater mean increase in heart rate (12.3 vs 3.6 bpm) and QT interval corrected for heart rate using the Bazett formula (QTcB) (10.5 vs 5.1 msec), consistent with the heart rate increase; mean change in QT interval corrected for heart rate using the Fridericia formula (QTcF) was similar for levomilnacipran ER and placebo patients (-1.3 msec vs 1.4 msec).
Suicidality. During the open-label treatment period, 14 percent of patients reported suicidal ideation based on assessment by the C-SSRS; 70 percent of suicidal ideation was in the least severe category of ideation (“wish to be dead”, with no intent to act). One patient had an aborted suicide attempt and continued in the study; two patients had actual attempts, which corresponded to SAEs, and led to study discontinuation. One suicide attempt occurred after 52 days of levomilnacipran ER treatment in a patient with a history of suicidal behavior; the other suicide attempt occurred after 32 days of levomilnacipran ER treatment in a patient with no history of suicidal behavior. None of the attempts were considered by the Investigator to be related to study drug.
No reports of suicidal behavior occurred during the double-blind treatment period. During the double-blind treatment period, the incidence of suicidal ideation was higher in the levomilnacipran ER group (4.8%) than in the placebo group (2.7%).
Discussion
This Phase III trial compared the efficacy of fixed-dose levomilnacipran ER (40, 80, 120mg/day) and placebo for the prevention of relapse in patients with MDD following response to a 12-week course of therapy with flexible-dose open-label levomilnacipran ER 40 to 120mg/day; safety and tolerability were also evaluated. Although time to relapse during double-blind treatment was prolonged in the levomilnacipran ER group compared with placebo, the treatment effect did not reach statistical significance. The relapse rates that were observed in this study (placebo=20.5%, levomilnacipran ER=13.9%) were lower than the relapse rates anticipated in the statistical analysis plan (placebo=38%, levomilnacipran ER=20%), which compromised the projected statistical power. As such, this study was underpowered and lacked assay sensitivity to demonstrate a difference between groups, making it more of a failed study than a negative study.
The lack of statistical difference between levomilnacipran ER and placebo in this study is inconsistent with the preponderance of evidence in the literature that demonstrates statistically significant reduction in time to relapse and relapse rates for antidepressants relative to placebo.[24–26] Relapse prevention trials in other antidepressants have typically shown placebo relapse rates on the order of 40 to 50 percent over 24 weeks after discontinuing active therapy; even with continuation of active treatment, relapse rates for antidepressants have been reported in the range of 20 to 30 percent.[8,27,28]
A large, pooled analysis of 31 relapse prevention trials24 found that the average rate of relapse was 41 percent for patients on placebo compared with 18 percent for patients on active treatment. Although the included studies were somewhat heterogeneous in design in terms of length of treatment before and after randomization, analysis of the studies that were most similar to the design of the current trial (i.e., 8 weeks active treatment/24-week follow-up after randomization) found an average rate of relapse of 34 percent for patients on placebo compared with 15 percent for patients on active treatment. In naturalistic follow-up studies, rates of depressive relapse following remission from an acute episode have been estimated to be 27 to 50 percent by six months.[29]
The lower than expected rates of relapse observed in the current study may also be related to the specific patient population that was recruited and enrolled, and/or aspects of the study design.
Although this study used an accepted depression relapse prevention paradigm in which responders to short-term antidepressant treatment are randomly assigned to double-blind continuation of the antidepressant or switch to placebo after gradual down-taper,[30] several unanticipated factors may have contributed to the results. For example, it has been demonstrated that it is more difficult to detect a treatment effect in studies enrolling patients with lower baseline severity of depression.[31–35] Of note here, patients with a low baseline MADRS threshold score (22 or greater) were allowed to participate in this levomilnacipran ER relapse trial, perhaps blunting the ability to assess relapse in this study.
Post-hoc analyses of patients who were more severely depressed at entry into the open-label phase of this study suggested that they were at greater risk of relapse if they were treated with placebo compared with levomilnacipran ER during double-blind treatment. These findings are consistent with a failed relapse prevention trial in patients randomized to the investigational agent agomelatine or placebo after response to eight weeks of open-label treatment with agomelatine.[36] While there was no statistically significant difference in time to relapse between agomelatine and placebo in the overall group during double-blind treatment, agomelatine was superior to placebo in preventing relapse in a subset of patients with a Hamilton Depression Rating Scale[37] baseline total score greater than 25, a commonly used threshold score to define severe depression.[38]
Partial versus complete remission of depressive symptoms has also been shown to be a powerful predictor of depression relapse risk.[4,39] In the current study, robust improvement in symptoms during open-label treatment may have produced a double-blind population with relatively low relapse risk. At the time of randomization, 87 percent of the population was in remission (MADRS total score of 10 or less), and mean double-blind baseline scores of 6 for MADRS total score and 2 for CGI-S (“borderline ill”) suggest relatively little residual depressive symptomatology.
In the current study, patients who responded to open-label treatment but had greater residual symptomatology (MADRS total score of 4 or greater) and a longer duration of MDD also seemed to be at greater risk for relapse when treated with placebo compared to continuation treatment with levomilnacipran ER. These findings are consistent with both controlled clinical trials and naturalistic studies that found that patients who were asymptomatic following recovery from a depressive episode remained free of relapse/recurrence for a prolonged period compared with patients who had residual symptoms.[39,40]
Rates of relapse in the current study were determined during the 24 weeks after randomization to placebo or levomilnacipran ER; numerical separation between active treatment and placebo became apparent beginning after approximately four weeks, with the difference appearing to increase after Week 20. Given the lower than expected overall rates of relapse in this study, using an alternative approach and continuing the trial until a predetermined number of relapse events had occurred might have detected a treatment effect. While an event-driven study design would likely increase the probability of detecting a treatment effect for an active medication, a trial of this nature may be longer in duration and more costly to complete. It could also lead to difficulty interpreting whether an observed effect is demonstrating the prevention of relapse of the index episode or a delay in the emergence of a new episode of depression.[3]
The protocol-specified criteria for relapse in this study were arguably more rigorous than those used in other depression relapse trials.[28,41–43] In some of these studies, relapse criteria could be met at a single visit, while in others the clinical judgment of the Investigator was included as a criterion. Anecdotal reports from Investigators participating in the current trial suggest that a number of patients failed to meet relapse criteria at two consecutive visits but nonetheless showed symptoms of a clinically worsening condition; declining clinical condition that did not meet relapse criteria may have contributed to our lower than expected overall rates. This view is supported by a placebo-controlled relapse prevention study of duloxetine, which demonstrated higher rates of relapse, especially for placebo-treated patients, via Investigator assessment compared with protocol-specified criteria.[8]
A final study design consideration that may have contributed to the lower than expected rate of relapse among placebo-treated patients was the 2:1 randomization in favor of levomilnacipran ER. The greater probability of receiving active treatment relative to placebo may have increased patients’ expectations of a continued therapeutic response.[44] Several recent short-term studies have demonstrated a correlation between lower probability of being treated with placebo and increased treatment response in the treatment of depression.[34,45] Whether this correlation holds in relapse prevention trials is worth further exploration.
While the current study failed to show a statistically significant benefit for levomilnacipran ER relative to placebo in the prevention of relapse in patients with MDD, there was strong response to 12 weeks of open-label levomilnacipran ER treatment. While open-label improvement is difficult to interpret due to the lack of a placebo comparator, this robust response may have inadvertently produced a double-blind treatment population that was largely in remission and had little residual depressive symptomatology. These results are consistent with results from the long-term open-label levomilnacipran ER study[18] that showed good retention and sustained MADRS improvements in patients who responded to eight-week double-blind treatment with levomilnacipran ER or placebo.
While a lack of statistical power may have compromised the ability to detect a between-group difference for levomilnacipran ER and placebo, lower than expected relapse rates and sustained open-label improvement are encouraging findings. Levomilnacipran ER was generally well tolerated in this study with common AEs that are typical of agents that inhibit both serotonin and norepinephrine reuptake;[46] the most common AEs often resolved over time, and no new safety findings were noted with long-term levomilnacipran ER treatment.
References
1. Rush AJ, Trivedi MH, Wisniewski SR, Nierenberg AA, Stewart JW, et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry. 2006;163(11):1905–1917.
2. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision. Washington DC: American Psychiatric Press Inc.; 2000.
3. Frank E, Prien RF, Jarrett RB, et al. Conceptualization and rationale for consensus definitions of terms in major depressive disorder. Remission, recovery, relapse, and recurrence. Arch Gen Psychiatry. 1991;48(9):851–855.
4. Keller MB. Past, present, and future directions for defining optimal treatment outcome in depression: remission and beyond. JAMA. 2003;289(23):3152–3160.
5. American Psychiatric Association. APA Practice Guideline for the Treatment of Patients With Major Depressive Disorder, 3rd ed. Arlington, VA: American Psychiatric Press Inc.,; 2010. http://psychiatryonline.org/content.aspx?bookid=28§ionid=1667485#657567.
6. Kocsis JH, Thase ME, Trivedi MH, et al. Prevention of recurrent episodes of depression with venlafaxine ER in a 1-year maintenance phase from the PREVENT Study. J Clin Psychiatry. 2007 Jul;68(7):1014–1023.
7. Kornstein SG, Bose A, Li D, et al. Escitalopram maintenance treatment for prevention of recurrent depression: a randomized, placebo-controlled trial. J Clin Psychiatry. 2006;67(11):1767–1775.
8. Perahia DG, Maina G, Thase ME, et al. Duloxetine in the prevention of depressive recurrences: a randomized, double-blind, placebo-controlled trial. J Clin Psychiatry. 2009;70(5):706–716.
9. Fetzima [package insert]. St. Louis, MO: Forest Pharmaceuticals, Inc; 2013.
10. Auclair AL, Martel JC, Assie MB, et al. Levomilnacipran (F2695), a norepinephrine-preferring SNRI: profile in vitro and in models of depression and anxiety. Neuropharmacology. 2013;70:338–347.
11. Deecher DC, Beyer CE, Johnston G, et al. Desvenlafaxine succinate: a new serotonin and norepinephrine reuptake inhibitor. J Pharmacol Exp Ther. 2006;318(2):657–665.
12. Sambunaris A, Bose A, Gommoll C, et al. A Phase III, double-blind, placebo-controlled, flexible-dose study of levomilnacipran ER in patients with major depressive disorder. J Clin Psychopharmacol. 2014;34(1):47–56.
13. Asnis G, Bose A, Gommoll C, et al. The efficacy and safety of levomilnacipran SR 40 mg, 80 mg, or 120 mg in major depressive disorder: a phase III, randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2013;74(3):242–248.
14. Bakish D, Bose A, Gommoll C, et al. Levomilnacipran ER 40 mg and 80 mg in major depressive disorder: a phase III, randomized, double-blind, fixed-dose, placebo-controlled study. J Psychiatry Neurosci. 2014;39(1):40–49.
15. Montgomery S, Mansuy L, Ruth A, et al. The efficacy and safety of levomilnacipran sustained release in moderate to severe major depressive disorder: a randomized, double-blind, placebo-controlled, proof-of-concept study. J Clin Psychiatry. 2013;74(4):363–369.
16. Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. Br J Psychiatry. 1979;134:382–389.
17. Gommoll C, Greenberg WM, Chen, C. A randomized, double-blind, placebo-controlled study of flexible doses of levomilnacipran ER (40–120 mg/day) in patients with major depressive disorder. J Drug Assessment. 2014;3:10–19.
18. Mago R, Forero G, Greenberg WM, et al. Safety and tolerability of levomilnacipran ER in major depressive disorder: results from an open-label, 48-week extension study. Clin Drug Investig. 2013;33(10):761–771.
19. Guy W. ECDEU Assessment Manual for Psychopharmacology. “Clinical Global Impressions.” Rockville, MD: National Institute of Mental Health; 218-222. DHEW Publication No. 76-338. 1976.
20. Sheehan DV, Lecrubier Y, Sheehan KH, Amorim P, Janavs J, et al. The Mini-International Neuropsychiatric Interview (M.I.N.I.): the development and validation of a structured diagnostic psychiatric interview for DSM-IV and ICD-10. J Clin Psychiatry. 1998;59(Suppl 20):22–33.
21. Posner K, Brown GK, Stanley B, et al. The Columbia-Suicide Severity Rating Scale: initial validity and internal consistency findings from three multisite studies with adolescents and adults. Am J Psychiatry. 2011;168(12):1266–1277.
22. Sheehan DV, Harnett-Sheehan K, Raj BA. The measurement of disability. Int Clin Psychopharmacol. 1996;11(Suppl 3):89–95.
23. Watkins PB, Seligman PJ, Pears JS, et al. Using controlled clinical trials to learn more about acute drug-induced liver injury. Hepatology. 2008;48(5):680–1689.
24. Geddes JR, Carney SM, Davies C, et al. Relapse prevention with antidepressant drug treatment in depressive disorders: a systematic review. Lancet. 2003;361(9358):653–661.
25. Hansen R, Gaynes B, Thieda P, et al. Meta-analysis of major depressive disorder relapse and recurrence with second-generation antidepressants. Psychiatr Serv. 2008;59(10):1121–1130.
26. Reid S, Barbui C. Long term treatment of depression with selective serotonin reuptake inhibitors and newer antidepressants. BMJ. 2010;340:c1468.
27. Reimherr FW, Amsterdam JD, Quitkin FM, et al. Optimal length of continuation therapy in depression: a prospective assessment during long-term fluoxetine treatment. Am J Psychiatry. 1998;155(9):1247–1253.
28. Simon JS, Aguiar LM, Kunz NR, Lei D. Extended-release venlafaxine in relapse prevention for patients with major depressive disorder. J Psychiatr Res. 2004;38(3):249–257.
29. Anderson IM, Ferrier IN, Baldwin RC, et al. Evidence-based guidelines for treating depressive disorders with antidepressants: a revision of the 2000 British Association for Psychopharmacology guidelines. J Psychopharmacol. 2008 Jun;22(4):343-396.
30. Nierenberg AA, Petersen TJ, Alpert JE. Prevention of relapse and recurrence in depression: the role of long-term pharmacotherapy and psychotherapy. J Clin Psychiatry. 2003;64(Suppl 15):13–17.
31. Khan A, Leventhal RM, Khan SR, Brown WA. Severity of depression and response to antidepressants and placebo: an analysis of the Food and Drug Administration database. J Clin Psychopharmacol. 2002;22(1):40–45.
32. Khin NA, Chen YF, Yang Y, et al. Exploratory analyses of efficacy data from major depressive disorder trials submitted to the US Food and Drug Administration in support of new drug applications. J Clin Psychiatry. 2011;72(4):464–472.
33. Khan A, Schwartz K, Kolts RL, et al. Relationship between depression severity entry criteria and antidepressant clinical trial outcomes. Biol Psychiatry. 2007;62(1):65–71.
34. Papakostas GI, Fava M. Does the probability of receiving placebo influence clinical trial outcome? A meta-regression of double-blind, randomized clinical trials in MDD. Eur Neuropsychopharmacol. 2009;19(1):34–40.
35. Fournier JC, DeRubeis RJ, Hollon SD, et al. Antidepressant drug effects and depression severity: a patient-level meta-analysis. JAMA. 2010;303(1):47–53.
36. Goodwin GM, Boyer P, Emsley R, et al. Is it time to shift to better characterization of patients in trials assessing novel antidepressants? An example of two relapse prevention studies with agomelatine. Int Clin Psychopharmacol. 2013;28(1):20–28.
37. Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960;23:56–62.
38. Nemeroff CB. The burden of severe depression: a review of diagnostic challenges and treatment alternatives. J Psychiatr Res. 2007;41(3–4):189–206.
39. Judd LL, Akiskal HS, Maser JD, et al. Major depressive disorder: a prospective study of residual subthreshold depressive symptoms as predictor of rapid relapse. J Affect Disord. 1998 Sep;50(2–3):97–108.
40. Zimmerman M, Chelminski I, Posternak M. A review of studies of the Montgomery-Asberg Depression Rating Scale in controls: implications for the definition of remission in treatment studies of depression. Int Clin Psychopharmacol. 2004;19(1):1–7.
41. Doogan DP, Caillard V. Sertraline in the prevention of depression. Br J Psychiatry. 1992;160:217–222.
42. Montgomery SA, Dunbar G. Paroxetine is better than placebo in relapse prevention and the prophylaxis of recurrent depression. Int Clin Psychopharmacol. 1993;8(3):189–195.
43. Robert P, Montgomery SA. Citalopram in doses of 20–60 mg is effective in depression relapse prevention: a placebo-controlled 6 month study. Int Clin Psychopharmacol. 1995;10(Suppl 1):29–35.
44. Rutherford BR, Roose SP. A model of placebo response in antidepressant clinical trials. Am J Psychiatry. 2013;170(7):723–733.
45. Sinyor M, Levitt AJ, Cheung AH, et al. Does inclusion of a placebo arm influence response to active antidepressant treatment in randomized controlled trials? Results from pooled and meta-analyses. J Clin Psychiatry. 2010;71(3):270–279.
46. Stahl SM, Grady MM, Moret C, Briley M. SNRIs: their pharmacology, clinical efficacy, and tolerability in comparison with other classes of antidepressants. CNS Spectr. 2005;10(9):732–747.