

by Muhammad Hassan Majeed, MD; Muhammad Ubaidulhaq, MD; Anesh Rugnath, MD; and Ike Eriator, MD
Dr. Majeed is Attending Psychiatrist with the Department of Psychiatry at Natchaug Hospital in Norwich, Connecticut. Dr. Ubaidulhaq is Pain Medicine Fellow with the Department of Anesthesiology at University of Mississippi Medical Center in Jackson, Mississippi. Dr. Rugnath is Attending Physician with the Department of Anesthesiology & Pain Medicine at University of Mississippi Medical Center in Jackson, Mississippi. Dr. Eriator is Chairman with the Department of Anesthesiology & Pain Medicine at University of Mississippi Medical Center in Jackson, Mississippi.
Funding: No funding was provided.
Disclosures: The authors have no conflicts of interest relevant to the content of this article.
Innov Clin Neurosci. 2018;15(11–12):33–35
Abstract: Pain insensitivity disorders are rare; however, when individuals are insensitive to pain, they are significantly more vulnerable to physical injuries, with higher morbidity and mortality rates, compared with the general population. The authors present the case of an 11-month-old male infant with SCN 9A gene mutation that resulted in congenital insensitivity to pain, while his mother, with a different mutation of the same gene, had hypersensitivity to pain. This is a rare familial presentation of the extreme ends of pain sensitivity, and might be the first such example in medical literature. There is little available information regarding the treatment of pain insensitivity disorders. The authors provide a brief discussion regarding diagnosis (including differentials), known etiology, and treatment of congenital insensitivity to pain, of which a multidisciplinary treatment approach is recommended.
Keywords: Congenital insensitivity to pain, CIP, SCN9A mutation, hereditary sensory and autonomic neuropathy Type IID, HSAN IID
Pain is the most common symptom reported in medical office visits and remains one of the most important symptoms in all clinical medicine.1 Clinicians who manage pain disorders rarely encounter patients with insensitivity to pain. However, such insensitivity makes a patient vulnerable to serious injuries due to the absence of protective reactions to noxious stimuli.2 Almost 20 percent of these patients die from hyperpyrexia before the age of three years.3 Patients with pain insensitivity require specific care to prevent life-limiting complications.
Congenital insensitivity to pain (CIP) is inherited in an autosomal recessive pattern. It is caused by mutation of the SCN9A gene located on chromosome 2q24.3.4 The SCN9A gene determines the formation of the sodium channels, which help in the conduction of action potential across excitable cells. Genetic polymorphism has been identified in the expression of the SCN9A gene leading to several pain phenotypes—from hypersensitivity to complete insensitivity.5 Different mutations in the SCN9A gene that encode voltage-gated sodium channels (Nav1.7) can cause primary erythermalgia, paroxysmal extreme pain disorder, and channelopathy-associated insensitivity to physical pain.6 These variants represent distinct forms of peripheral neuronal sodium channelopathies. CIP is caused by balletic null mutations that result in loss of Nav 1.7 function, which increases the transcription of messenger ribonucleic acid (mRNA) and higher levels of enkephalins in sensory neurons. In patients with these mutations, nociceptive stimuli, such as temperature, pressure, and pinpricks, do not elicit a pain response.2
With consent of the parents, we present a case of hereditary sensory and autonomic neuropathy Type IID (HSAN IID) in an 11-month old male infant due to mutations in the SCN9A gene.7 This autosomal recessive trait is present on chromosome 2q 24.3. The mother of the child had the same gene mutation with a completely opposite manifestation—hypersensitivity to pain. She had an activating mutation of SCN9A (Nav1.7) on chromosome 2. To our knowledge, this familial variant of extreme pain perception has not yet been described in the literature.
Case Presentation
An 11-month-old Caucasian male infant was referred to the neurology department of the University of Missouri Medical Center in Jackson, Mississippi, by his primary care physician due to heightened pain tolerance. The patient did not cry, flinch, or whine during circumcision or immunization. Rather, he smiled and reacted as though he was being tickled on such occasions. At approximately six months of age, his parents noticed that he would chew and bite his own tongue and that he had recurrent fevers without sweating. At nine months of age, chewing on the extensor halluces longus produced bone exposure on the left hallux.
On examination, the patient’s vital signs were stable. His head circumference was at the 50th percentile, length was at the 85th percentile and weight was greater than the 97th percentile. He displayed second and third toe syndactyly; multiple bite marks on his tongue, lips, and appendages; and subtle hypotonia, but no weakness (Figure 1). An age-appropriate Bibinski sign was elicited. The sensory examination was limited, but the patient did not exhibit any signs of discomfort from firm pressure on his toes or fingertips. His parents indicated that he did not react to hot or cold surfaces. Because of his age, we could not assess anosmia. The parents also reported that their son had symptoms of anhidrosis. The patient’s mother reported her own history of second and third toe syndactyly and allodynia. The patient’s father and maternal grandfather reportedly also had high tolerance to pain. Differential diagnoses included child abuse, Lesch-Nyhan Syndrome, adrenoleukodystrophy, toxic epidermal necrolysis, and CIP. Genetic testing was discussed with the parents and, upon their consent, was conducted.
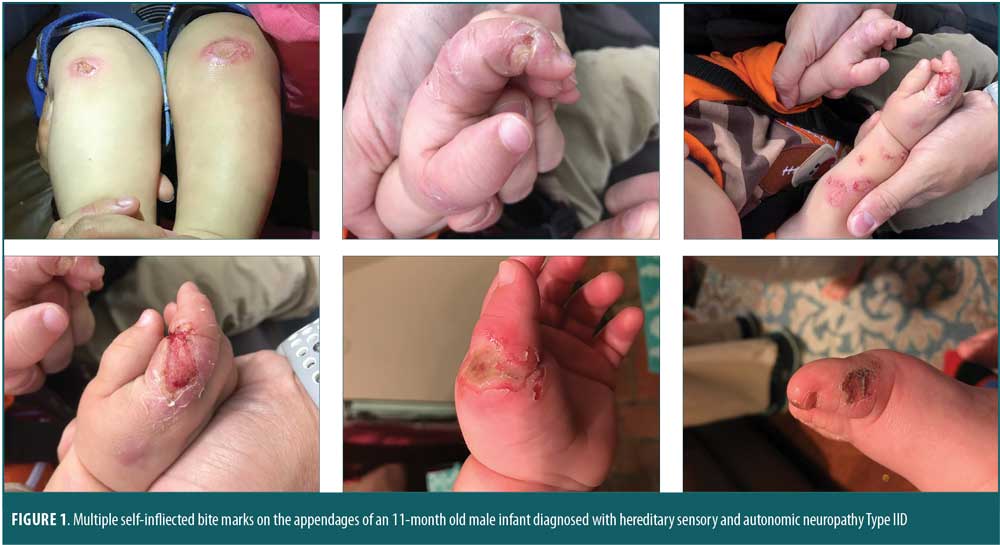
Upon genetic analysis, chromosomal microarray showed a 67 kb likely pathogenic loss at 2q24.3 within the SCN9A gene. Sequencing of the SCN9A gene revealed p.Ser 1387Phe (S1387F) (TCT>TTT): C.4160 C>T in exon 22 of the SCN9A gene, which is classified as a “heterozygous variant of uncertain significance.” Other routine investigations, including hypoxanthine-guanine phosphoribosyl-transferase and uric acids levels, were within normal ranges. Quantitative amino acids, very long chain fatty acid, urine organic acids, and a fragile X deoxyribonucleic acid (DNA) probe were reported to be within normal limits. Electromyography findings showed a reduction in the sensory compound action potential and increased latencies in all the nerves, which was reported as possible axonal sensory peripheral neuropathy. A magnetic resonance imaging (MRI) scan of the brain, with and without contrast, was normal.
Discussion
Diagnosis. History, clinical examination, labs, and other investigative measures indicated a working diagnosis of HSAN IID in our patient. Patients with inactive mutations in SCN9A typically are of normal intelligence, present with CIP and congenital anosmia, and do not show signs of autonomic function loss. A subset of these patients has HSAN IID, which presents with CIP and autonomic function loss, evident by anhidrosis and a variable presentation of anosmia.5
HSAN IID is characterized by progressively reduced sensation to pain, temperature, and touch, which, in turn, can cause unintentional injury, including bruising, wounds, auto-amputation of the digits, osteomyelitis, and other complications.7
Parents or caregivers are usually the first to observe signs of HSAN IID when their child does not show signs of pain after injuries or with the needle prick for immunizations. The symptoms of the disorder typically become more pronounced with the eruption of teeth, which can cause more severe injury when the child bites his or her own tongue, lips, and/or other parts of the body.2 Families might be suspected of child abuse or neglect due to the presence of severe bruising, wounds, and/or scars present on the child’s body.
Etiology. Sensory neurons transmit acute and chronic pain signals. Nociceptors are sensory neurons that transmit signals when there is a real or perceived tissue injury from mechanical force, variation of temperature, or when the body comes in contact with chemicals.8 These nociceptors have voltage-gated sodium channels (Nav) that play a pivotal role in the generation of action-potential in these cells. Voltage-gated sodium channels play a vital role in converting mechanical and chemical stimuli into electrical signals within excited cells. So far, nine different channels encoded by nine different genes (SCN1-9A) have been recognized. Nav1.7 channel is a protein made of 1.977 amino acids, which form four similar domains, each of which comprises six transmembrane regions.2 An important sub-type of the sodium channel is Nav17, which determines the threshold for the excitement of nociceptors and is also involved in the amplification of these stimuli.9 The SCN9A gene encodes for Nav1.7 channels, which are associated with insensitivity to pain in the case of inactivating mutation, and the opposite phenotype, hyperalgesia, in the case of activating mutations.10 Nav1.7 channels are prominent in the olfactory system and might play a role in the regulation of neurotransmitters in the central nervous system.11
In CIP, the sensory and autonomic neurons either do not develop properly or degenerate at an early age.12 Lack or mutation of several nerve growth factors are pathophysiological mechanisms involved in CIP, which can lead to degeneration and atrophy of sensory neurons.13 Sometimes only the functioning is disrupted, with the structural integrity of neurons preserved.14 In our patient, the S1387F variant, a non-conservative amino acid substitution, likely impacted secondary protein structures. This substitution occurs between the S5 and S6 transmembrane segments of the third homologous domain.
Treatment. Currently, there is no known cure or gene therapies available for CIP. The most common causes of mortality are injuries and subsequent infections and fever.15 Treatment is focused on prevention and active management of the symptoms, and for that, a good therapeutic alliance with a family member is important. Genetic counseling is an integral part of the prevention. In utero, genetic sampling of amniotic fluid or chronic villus sampling can also detect a genetic mutation in the fetus. A pre-implantation genetic diagnosis can provide a choice to the parents of implanting only healthy embryos in the uterus.16
Naloxone. Opioid-antagonist naloxone has shown promising results in treatments reversing the analgesia caused by mutation of SCN9A gene.17 With a loss of Nav 1.7 expression, there is an upregulation of an endogenous opioid system leading to insensitivity to pain. Naloxone increases noxious input to the central nervous system and significantly reduces analgesia in CIP. Research has found that naloxone can benefit both thermal and mechanical pain states.
Carbamazepine and gabapentin. There is some evidence that carbamazepine and gabapentin might help with SCN9A mutation variants of paroxysmal extreme pain disorder, but there is no information available regarding its use in pain insensitivity disorders.18 In contrast, a novel drug that can selectively block selective Nav channels in nociceptors could be the Holy Grail for effectively treating pain without risk of abuse and addiction.19
Patient/caregiver education. Patients and their families should be educated on behavior skills to prevent injuries and promote early detection of wounds or illnesses. Children might need physical and occupation therapy to learn certain motor skills to manage their lives. Ever increasing knowledge and understanding of genetic and molecular models of pain will eventually translate into better treatment for genetic pain syndromes. Meanwhile, these patients need multidisciplinary care to improve the quality of their lives and to prevent accidental injuries or death.
Summary
Different mutations of the SCN9A gene that encode voltage-gated sodium channels (Nav1.7) can cause neuronal sodium channelopathies. Patients with the loss of function mutation can present with congenital insensitivity to pain. Naloxone has shown some promising initial results in the treatment of pain insensitivity, but these findings need to be replicated. Genetic counseling, planning, and safety assessment are important to prevent injuries. A multi-disciplinary team approach is necessary to treat these patients.
References
- Gureje O, Von Korff M, Simon GE, Gater R. Persistent pain and well-being: a World Health Organization Study in Primary Care. JAMA. 1998;280(2):147–151.
- Bennett DLH, Woods CG. Painful and painless channelopathies. Lancet Neurol. 2014;13(6):587–599.
- Rosemberg S, Marie SK, Kliemann S. Congenital insensitivity to pain with anhidrosis (hereditary sensory and autonomic neuropathy type IV). Pediatr Neurol. 1994;11(1):50–56.
- Cox JJ, Reimann F, Nicholas AK, et al. An SCN9A channelopathy causes congenital inability to experience pain. Nature. 2006;444(7121):894–898.
- Drenth JPH, Waxman SG. Mutations in sodium-channel gene SCN9A cause a spectrum of human genetic pain disorders. J Clin Invest. 2007;117(12):3603–3609.
- Reilly MM, Shy ME. Diagnosis and new treatments in genetic neuropathies. J Neurol Neurosurg Psychiatry. 2009;80(12):
1304–1314. - Kurth I. Hereditary Sensory and Autonomic Neuropathy Type II. GeneReviews(®). University of Washington, Seattle; 1993.
- Reichling DB, Green PG, Levine JD. The fundamental unit of pain is the cell. Pain. 2013;154:S2–9.
- Cummins TR, Howe JR, Waxman SG. Slow closed-state inactivation: a novel mechanism underlying ramp currents in cells expressing the hNE/PN1 sodium channel. J Neurosci. 1998;18(23):9607–9619.
- Kurban M, Wajid M, Shimomura Y, Christiano AM. A nonsense mutation in the SCN9A gene in congenital insensitivity to pain. Dermatology. 2010;221(2):179–183.
- Minett MS, Nassar MA, Clark AK, et al. Distinct Nav1.7-dependent pain sensations require different sets of sensory and sympathetic neurons. Nat Commun. 2012;3(1):791.
- Rotthier A, Baets J, Timmerman V, Janssens K. Mechanisms of disease in hereditary sensory and autonomic neuropathies. Nat Rev Neurol. 2012;8(2):73–85.
- Indo Y, Tsuruta M, Hayashida Y, et al. Mutations in the TRKA/NGF receptor gene in patients with congenital insensitivity to pain with anhidrosis. Nat Genet. 1996;13(4):485–488.
- Rotthier A, Baets J, Vriendt ED, et al. Genes for hereditary sensory and autonomic neuropathies: a genotype-phenotype correlation. Brain. 2009;132(10):2699–2711.
- Sasnur AH, Sasnur PA, Ghaus-Ul RSM. Congenital insensitivity to pain and anhidrosis. Indian J Orthop. 2011;45(3):269–271.
- Dolan SM, Goldwaser TH, Jindal SK. Preimplantation genetic diagnosis for mendelian conditions. JAMA. 2017;318(9):859.
- Minett MS, Pereira V, Sikandar S, et al. Endogenous opioids contribute to insensitivity to pain in humans and mice lacking sodium channel Nav1.7. Nat Commun. 2015;6:8967.
- Natkunarajah J, Atherton D, Elmslie F, et al. Treatment with carbamazepine and gabapentin of a patient with primary erythermalgia (erythromelalgia) identified to have a mutation in the SCN9A gene, encoding a voltage-gated sodium channel. Clin Exp Dermatol. 2009;34(8):e640–642.
- Emery EC, Luiz AP, Wood JN. Na v 1.7 and other voltage-gated sodium channels as drug targets for pain relief. Expert Opin Ther Targets. 2016;20(8):975–983.