
 by Fatemeh Naddafi, MSc; Mohsen Reza Haidari, PhD; Gholamreza Azizi, MSc; Reza Sedaghat, PhD; and Abbas Mirshafiey, PhD
by Fatemeh Naddafi, MSc; Mohsen Reza Haidari, PhD; Gholamreza Azizi, MSc; Reza Sedaghat, PhD; and Abbas Mirshafiey, PhD
Ms. Naddafi is from Department of Cellular and Molecular Biology, Kish International Campus, University of Tehran, Tehran, Iran; Dr. Haidari is from Department of Neurology, Faculty of Medicine, Baqiyatallah University of Medical Sciences, Tehran, Iran; Mr. Azizi is from Imam Hassan Mojtaba Hospital, Alborz University of Medical Sciences, Karaj, Iran; Dr. Sedaghat is from Department of Anatomy and Pathology, Faculty of Medicine, Shahed University, Tehran, Iran; and Dr. Mirshafiey is from Department of Immunology, School of Public Health, Tehran University of Medical Sciences, Tehran, Iran.
Innov Clin Neurosci. 2013;10(4):20–25
Funding: No funding was provided for the preparation of this article.
Financial Disclosures: The authors do not have conflicts of interest relevant to the content of this article.
Key Words: Multiple sclerosis, nicotine, MMP-2
Abstract: Background: Multiple sclerosis is an autoimmune, neurodegenerative disease of the central nervous system. The cause of multiple sclerosis is still unknown, and there is no cure for multiple sclerosis. Experimental autoimmune encephalomyelitis is considered as an animal model for multiple sclerosis. The therapeutic role of nicotine has been proven to be effective in both Alzheimer’s and Parkinson’s disease, thus we examined, for the first time, the role of nicotine in the experimental autoimmune encephalomyelitis model.
Methods: Experimental autoimmune encephalomyelitis induction was performed according to Guang-Xian Zhang et al. Treatment with nicotine was started on Day 7 post-immunization. Prevention with nicotine was started on Day 7 pre-immunization. Also for in-vitro analysis, we used U-87 MG cell line to evaluate the inhibitory effect of nicotine in cell proliferation, pro-inflammatory cytokines (TNF-alpha, IL-1beta, IL-6) and MMP-2 activity by MTT, ELISA, and zymoanalysis methods, respectively. Moreover, the brains of mice were removed for histological analysis.
Results: Our findings showed that treatment with nicotine caused a significant reduction in the severity and onset of the experimental autoimmune encephalomyelitis. Histological analysis indicated that there was very mild and mild plaque in the brain sections of nicotine prevention and treatment groups, respectively.
Conclusion: Our data indicate that nicotine can significantly improve the clinical score and attenuate the demyelinating pathology typically found in experimental autoimmune encephalomyelitis, indicating that nicotine has protective effects in experimental model of multiple sclerosis.
Introduction
Multiple sclerosis (MS) is an autoimmune, inflammatory disease that is characterized by the breakdown of the blood-brain barrier (BBB) and accumulation of inflammatory cytokines infiltrate in the central nervous system (CNS).[1] Experimental autoimmune encephalomyelitis (EAE) is considered one of the best animal models for human autoimmune diseases, which gives insight into a variety of immunological and histopathological mechanisms of the CNS and for studying the therapeutic principles for preventing or curing the disease.[2] CD4+ T cells modulate the immune pathology by producing cytokines that activate cells of the innate immune system, such as macrophages.[3] Activated macrophages and microglia can secrete a broad amount of toxic mediators, including neuroactive cytokines, reactive oxygen species (ROS), and matrix metalloproteinases (MMPs). Furthermore, IL-17 is proven to disrupt the integrity of the BBB via the production of ROS within endothelial cells, ultimately leading to down-regulation of tight junction molecules.[4]
Nicotine is a known agonist of nicotinic acetylcholine receptors (nAChRs), members of the Cys-loop ligand-gated ion channel (LGIC) family that can respond to ACh as their endogenous ligand.[5] Acetylcholine (ACh) is known as the neurotransmitter in the cholinergic nervous system; however, it has been indicated that ACh is also produced by choline acetyltransferase (ChAT) in T lymphocytes. Moreover, it has been demonstrated that both T- and B-lymphocytes can express muscarinic and neuron type nicotinic ACh receptors (mAChRs and nAChRs, respectively).[6] It has been proven that nicotine exposure partially can decrease lymphocyte infiltration into the CNS, inhibit auto-reactive T cell proliferation and helper T cell cytokine production, down-regulate costimulatory protein expression on myeloid cells, and increase both the differentiation and recruitment of regulatory T cells, even in the absence or alpha7-nAChRs.[7] Recent studies indicate that nicotine does not restore integrity or function once dopaminergic neurons have been damaged, but it can attenuate ongoing neurodegenerative processes. Parkinson’s disease (PD) is a progressive neurodegenerative disease. The results from experimental animal models prove that nicotine may decrease PD progression.[8] Nicotinic receptor stimulation has a major role in several CNS functions, such as locomotor activity and cognition. Nicotine controls activity under physiological conditions as well as ameliorates locomotor deficits reported after nigrostriatal degeneration in both animals and humans.[9] The non-neuronal alpha7-nicotinic cholinergic receptors can be a primary target for nicotine through the JAK2 and STAT3/NF-kappaB pathways, which ultimately modulate the inhibition of pro-inflammatory gene transcription.[10] Although alpha7-receptors are considered major targets for neuroprotection, some data indicate the role of other receptor subtypes, including alpha4beta2 or alpha3beta4 in the neuroprotection process.[11] Nicotine can increase Treg-mediated immune suppression of lymphocytes through alpha7 nAChR. The effect is connected to the up-regulation of cytotoxic T-lymphocyte-associated antigen (CTLA-4) and Foxp3 expression and decreased IL-2 production in CD4+CD25+Tregs/ CD4+CD25- T-cell coculture supernatants. The alpha7nAChR can be identified as a key regulator for immunosuppressive function of CD4+CD25+Tregs.[12]
Materials and Methods
Animal selection and grouping. Six to 8 week old female wild-type (WT) C57BL/6 mice (n=/10 each group) weighing 16g were purchased from Pasteur Institute and maintained at the animal facilities of Tehran University, Iran. The animals were housed in standard conditions: constant temperature (22°C), humidity (relative, 25%), a 12-hour light/12-hour dark cycle, and were allowed free access to food and water. All animal experiments complied with the institutional guidelines.[13,14] There were three groups: control (patient), prevention, treatment.
Induction of EAE. MOG35–55/CFA Emulsion PTX 5X was purchased from Hooke Laboratories (USA). C57BL/6 mice developed chronic paralysis after immunization with MOG35–55 emulsified in Complete Freund’s Adjuvant (CFA). Mice developed EAE 15 days after immunization and stayed chronically paralyzed within the experiment (typically, mice were observed for 30–40 days).The kit consisted of two components, each delivered in separate pre-filled syringes—antigen (MOG35–55) in CFA emulsion and pertussis toxin (PTX) in PBS.[15]
Nicotine and therapeutic protocol. The 200?g/mL nicotine hydrogen tartrate salt (Sigma Aldrich, St. Louis, Missouri) and 2% saccharin (w/v) (Sigma Aldrich, St. Louis, Missouri) or pH-matched 2% saccharin (w/v) and 0.2% (v/v) tartaric acid (Sigma Aldrich, St. Louis, Missouri) were given to mice in drinking water as their sole source of fluid. Drinking solutions were changed twice weekly.[16] Prevention with nicotine was started on Day 7 pre-immunization and continued two weeks post-immunization.[17] Treatment with nicotine was started on Day 7 post-immunization and continued for three weeks.[17]
Clinical assessment of EAE. Clinically, animals were scored as follows: 1, limp tail; 2, partial hind leg paralysis; 3, complete hind leg paralysis or partial hind and front leg paralysis; 4, complete hind and partial front leg paralysis; 5, complete hind and partial front leg paralysis and reduced responsiveness to external stimuli. Mice were euthanized immediately if they scored 5 or if they scored 4 two days in a row.[18]
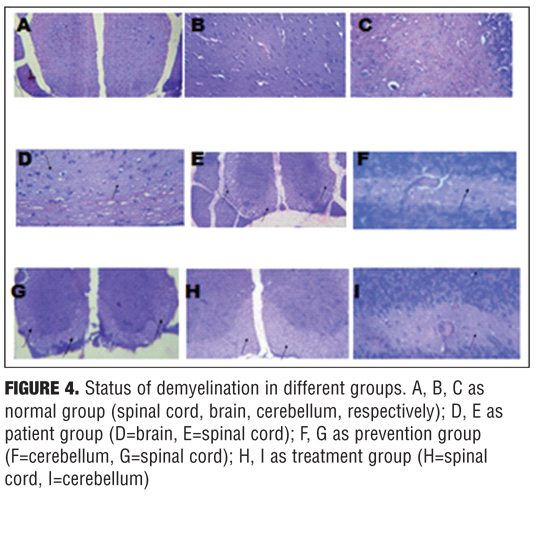
Histopathology. Brain, spinal cords, and cerebellum from treatment, prevention, and control mice were dissected 35 days after immunization with MOG35–55, fixed in 10% buffered formalin, and embedded in paraffin. For each animal, four equidistant coronal sections through the brain, four equidistant coronal sections through the spinal cord, and four equidistant coronal sections through the brainstem/cerebellum were selected and stained with Hematoxylin and Eosin (H&E) and Luxol Fast Blue (LFB). These methods were used to assess myelin loss.[14]
Cell culture and cell growth assessment. Human glioblastoma (U87MG) cell was purchased from Pasteur Institute and seeded at 5×103 cells per well on a 96-well plate. Nicotine was diluted in PBS and added at various concentrations (1, 5, 10, 50, 100, 500, and 1000?g/well) after four-hour cell attachment. Cells were incubated for 48 hours at 37°C and then removed from the medium. The number of viable cells was determined by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay (Sigma, St. Louis, Missouri): 200?l of MTT (0.5mg/mL in tissue culture medium without phenol red) were added and the plates were incubated for 2 to 4 hours. The medium was removed and 200?l of DMSO was added to each well to solubilize the formazan dye. The plates were read on an ELISA reader using the following settings: test wavelength 570nm, reference wavelength 630nm.[19]
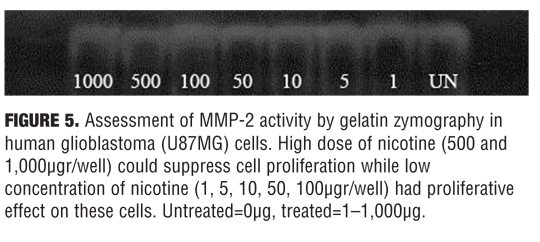
Evaluation of MMP-2 activity by zymoanalysis. The method of Kleiner and Stetler-Stevenson (1994) with slight modifications was used for evaluating the gelatinase (collagenase type IV or MMP-2) activity in conditioned media. Briefly, aliquots of human glioblastoma (U87MG) cells supernatant were subjected to electrophoresis in 2mg/mL gelatin type A- and sodium dodecyl sulfate (SDS)-containing polyacrylamide gels, under denaturing but nonreducing conditions, at a constant voltage of 80V. After electrophoresis, gels were gently washed three times in 2.5% Triton X-100 solution to remove SDS. They were then incubated at 37°C overnight in gelatinase activation buffer (0.1M Tris-HCl, pH 7.4, 10mM CaCl2) and subsequently stained with 0.5% Coomassie Brilliant Blue. After intensive destaining, proteolysis areas appeared as clear bands against a blue background. Quantitative evaluation of band intensity, on the basis of gray levels, was performed using the Molecular Analyst Software (BioRad, Hercules, California). Gelatinolytic activity was expressed in arbitrary units, relative to control samples. In all cases, data were normalized per the number of cells in each experimental condition, as determined by MTT assay performed after medium removal.[20]
C6 rat astrocyte treatment. The C6 rat astrocyte cell lines were seeded at a density of 1×104 cells per well in 96-well plates with 1µg/well of LPS (Sigma, USA), in the presence (10,100,500µM/well) or absence of nicotine. Cells were incubated for 48 hours, and culture supernatants were then collected and, after particulates were removed by centrifugation, stored in aliquots at 20?C until use.[21]
Quantification of TNF-alpha, IL-1beta, and IL-6. The levels of pro-inflammatory cytokines were determined in the supernatants of C6 cell cultures by a sandwich enzyme-linked immunosorbent assay (ELISA) technique. To evaluate TNF-alpha, we used ID Labs Kit (ID Labs inc. London, Canada). The amount of IL-1-beta and IL-6 was determined using kits from Boster (Boster Biological Technology, Ltd, USA). All assays were performed according to the manufacturers’ instructions. Absorbance was read at 450nm in a 96-well microplate ELISA reader.21
Statistical analysis. Statistical analysis was performed with Mann-Whitney U-test for nonparametric data and the differences in the clinical severity of EAE following MOG35-55-induction between the control and treatment and prevention mice groups were evaluated daily using the Kruskal–Wallis test. A p-value of 0.05 or less was considered statistically significant.[22]
Results
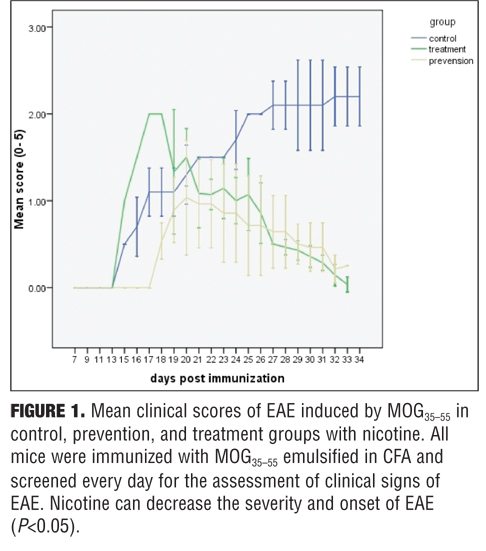
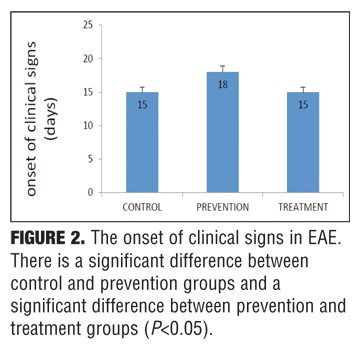
Clinical findings. The control group developed severe clinical manifestations, starting on Day 15, increasing in severity by Days 20 to 29, and maintained until sacrifice on Day 35. All animals were affected, and complete hind limb paralysis was observed in most animals. In contrast, the treatment group indicated mild and delayed clinical signs and the prevention group showed very mild clinical scores (Figure 1). Nicotine also delayed the onset of EAE in the prevention group (Figure 2).
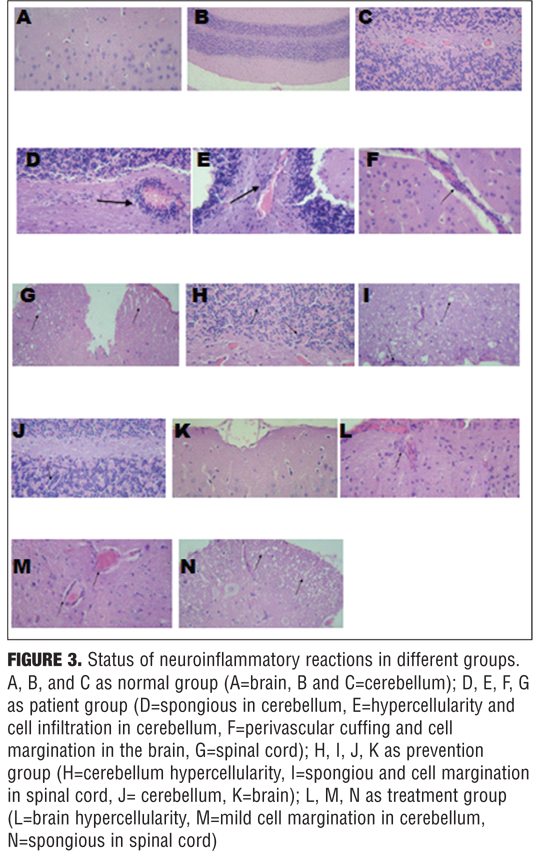
Histology findings. To evaluate the effects of nicotine treatment on inflammation and demyelination, brain, spinal cord, and cerebellum were analyzed by histological methods using light microscopy, 35 days after immunization. H&E and LFB staining showed marked multifocal and lymphohistiocytic inflammation (with perivascular and diffuse form) and myelin loss in the white matter of control group, especially around the inflamed areas. In contrast, in most sections from nicotine treatment group, there was less inflammation, and myelin sheets were preserved. Moreover, in most sections from prevention group, there was less inflammation than treatment group (p value ?0.05) (Figures 3 and 4).
Effect of nicotine on human glioblastoma (U87MG) cell. Human glioblastoma (U87MG) cells were exposed to a gradient of various concentrations of nicotine (1, 5, 10, 50, 100, 500, and 1000?g/well). High dose of nicotine (500 and 1000?gr/well could suppress cell proliferation while a low concentration of nicotine (1, 5, 10, 50, 100?g/well) could show proliferative effect on cell numbers.
Effect of nicotine on MMP-2 activity. Dose-response analysis of nicotine on MMP-2 activity is presented in Figure 5. High dose of nicotine (500 and 1000?gr/well) can suppress cell proliferation while low concentration of nicotine (1, 5, 10, 50, 100 ?g/well) had proliferative effect on these cells.
 MTT finding. MTT assay was used to evaluate the cell proliferation and neuronal cytotoxicity. The number of cells was analyzed by the MTT method. Following the treatment with nicotine, a decline of MTT reduction in U87MG cells was indicated with a dose-dependent manner, but the significant decrease can be only detected in the treatments with concentrations of 500 and 1000?g nicotine (data not shown).
MTT finding. MTT assay was used to evaluate the cell proliferation and neuronal cytotoxicity. The number of cells was analyzed by the MTT method. Following the treatment with nicotine, a decline of MTT reduction in U87MG cells was indicated with a dose-dependent manner, but the significant decrease can be only detected in the treatments with concentrations of 500 and 1000?g nicotine (data not shown).
The effect of nicotine on pro-inflammatory cytokines. We found that the 500µg/well of nicotine could decrease TNF-alpha production and the 100µg/well of nicotine suppressed IL-6 production. IL-1-beta level did not differ significantly.
Discussion
The neuroprotective effect of nicotine is proved by both the up-regulation of nicotinic receptors and the disruption of preformed beta-amyloid fibril (fA-beta) in AD.[23] Nicotine modulates some of its therapeutic actions in PD and AD through antioxidant mechanisms, whereas the animal experiments showed that nonantioxidant mechanisms are primarily responsible for nicotine’s multiplicity of motor/cognitive actions.[24] PD is a progressive neurodegenerative disorder, and the results from experimental animal models indicate that nicotine can decrease PD progression. Recent studies demonstrate that nicotine is not neurorestorative against nigrostriatal damage but it can attenuate ongoing neurodegenerative processes.[8] In addition, epidemiological evidence indicated that nicotine has beneficial and protective property in some neurodegenerative diseases such as AD. The complex I respiratory chain generates superoxide anion, and nicotine could inhibit ROS generation on rat brain mitochondria. Nicotine can bind to complex I of the respiratory chain and inhibit the NADH-ubiquinone reductase activity.[25] Moreover, nicotine prevents activation of NF-kappa-B (nuclear factor kappa-light-chain-enhancer of activated B cells) and c-Myc by inhibiting the activation of MAP kinases. In fact, nicotine decreases A-beta by the activation of a7nAChRs via MAPK, NF-kappa-B, and c-myc pathways. Nicotinic cholinergic receptor stimulation also induces neuroprotection against glutamate cytotoxicity by its inhibitory action on NO-formation and consequently, the activity of iNOS and the production of NO are down-regulated.[26] Due to the proven therapeutic effect of nicotine on AD and PD, we decided to study the role of nicotine in EAE as an animal model of MS. Our treatment group showed less inflammation in histopathological evaluation along with myelin sheet protection. Moreover, prevention group showed less inflammation compared with treatment group. Thus, nicotine might be recommended as a promising drug for MS therapy.
References
1. Bennett J, Basivireddy J, Kollar A, et al. Blood–brain barrier disruption and enhanced vascular permeability in the multiple sclerosis model EAE. J Neuroimmunol. 2010;229:180–191.
2. Kuerten S, Angelov D. Comparing the CNS morphology and immunobiology of different EAE models in C57BL/6 mice: a step towards understanding the complexity of multiple sclerosis. Ann Anat. 2008;190:1-15.
3. Kuerten S, Rottlaender A, Rodi M, et al. The clinical course of EAE is reflected by the dynamics of the neuroantigen-specific T cell compartment in the blood. Clin Immunol. 2010;137:422–432.
4. Kapadia M, Sakic B. Autoimmune and inflammatory mechanisms of CNS damage. Prog Neurobiol. 2011;95:301–333.
5. Dome P, Lazary J, Kalapos MP, Rihmer Z. Smoking, nicotine and neuropsychiatric disorders. Neurosci Biobehav Rev. 2010;34:295–342.
6. Kimura R, Ushiyama N, Fujii T, Kawashima K. Nicotine-induced Ca2+ signaling and down-regulation of nicotinic acetylcholine receptor subunit expression in the CEM human leukemic T-cell line. Life Sci. 2003;72:2155–8.
7. Hao J, Simard AR, Turner GH, et al. Attenuation of CNS inflammatory responses by nicotine involves alpha-7 and non-alpha-7 nicotinic receptors. Exp Neurol. 2011;227:110–119.
8. Quik M, Huang LZ, Parameswaran N, et al. Multiple roles for nicotine in Parkinson’s disease. Biochem Pharmacol. 2009;78:677–685.
9. Quik M, Jeyarasasingam G. Nicotinic receptors and Parkinson’s disease. Eur J Pharmacol. 2000;393:223–230.
10. Filippini P, Cesario A, Fini M, et al. The yin and yang of non-neuronal alpha-7-nicotinic receptors in inflammation and autoimmunity. Curr Drug Targets. 2012;13:644–655.
11. Egea J, Rosa AO, Sobrado M, et al. Neuroprotection afforded by nicotine against oxygen and glucose deprivation in hippocampal slices is lost in alpha-7 nicotinic receptor knockout mice. Neuroscience. 2007;145:866-72.
12. Wang DW, Zhou RB, Yao YM, et al. Stimulation of alpha-7 nicotinic acetylcholine receptor by nicotine increases suppressive capacity of naturally occurring CD4+CD25+ regulatory T cells in mice in vitro. J Pharmacol Exp Ther. 2010;335:553–561.
13. Kuerten S, Kostova-Bales DA, Frenzel LP, et al. MP4- and MOG:35–55-induced EAE in C57BL/6 mice differentially targets brain, spinal cord and cerebellum. J Neuroimmunol. 2007;189:31–40.
14. Yossi Gilgun-Sherki, Hana Panet, VeredHoldengreber, RonitMosberg-Galili, Daniel Offen. Axonal damage is reduced following glatiramer acetate treatment in C57/bl mice with chronic-induced experimental autoimmune Encephalomyelitis. Neurosci Res. 2003;47:201-7.
15. Mendel I, Kerlero de Rosbo N, Ben-Nun A. A myelin oligodendrocyte glycoprotein peptide induces typical chronic experimental autoimmune encephalomyelitis in H-2b mice: fine specificity and T cell receptor V beta expression of encephalitogenic T cells. Eur J Immunol. 1995;25:1951–1959.
16. Heath CJ, Horst NK, Picciotto MR. Oral nicotine consumption does not affect maternal care or early development in mice but results in modest hyperactivity in adolescence. Physiol Behav. 2010;101:764–769.
17. Quik M, O’Neill M, Perez XA. Nicotine neuroprotection against nigrostriatal damage: importance of the animal model. Trends Pharmacol Sci. 2007;28:229–235.
18. Thakker P, Leach MW, Kuang W, et al. IL-23 Is critical in the induction but not in the effector phase of experimental autoimmune encephalomyelitis. J Immunol. 2007;178:2589–2598.
19. Das A, Banik NL, Ray SK. N-(4-Hydroxyphenyl) retinamide induced both differentiation and apoptosis in human glioblastoma T98G and U87MG cells. Brain Res. 2008;1227:207–215.
20. Shariftabrizi A, Nifli AP, Ansari M, et al. Matrix metalloproteinase 2 secretion in WEHI 164 fibrosarcoma cells is nitric oxide-related and modified by morphine. Eur J Pharmacol. 2006;530:33–39.
21. Berghmans N, Dillen C, Heremans H. Exogenous IL-12 suppresses experimental autoimmune encephalomyelitis (EAE) by tuning IL-10 and IL-5 levels in an IFN-?-dependent way. J Neuroimmunol. 2006;176:63–75.
22. Roscoe WA, Welsh ME, Carter DE, Karlik SJ.VEGF and angiogenesis in acute and chronic MOG(35–55) peptide induced EAE. J Neuroimmunol. 2009;209:6–15.
23. Ono K, Hasegawa K, Yamada M, Naiki H. Nicotine Breaks Down Preformed Alzheimer’s beta-amyloid fibrils in vitro. Biol Psychiatry. 2002;52:880–886.
24. Linert W, Bridge MH, Huber M, et al. In-vitro and in-vivo studies investigating possible antioxidant actions of nicotine: relevance to Parkinson’s and Alzheimer’s diseases. Biochim Biophys Acta. 1999;1454:143–152.
25. Cormier A, Morin C, Zini R, et al. In-vitro effects of nicotine on mitochondrial respiration and superoxide anion generation. Brain Res. 2001;900:72–79.
26. Shimohama S, Akaike A, Kimura J. Nicotine-induced protection against glutamate cytotoxicity. Nicotinic cholinergic receptor-mediated inhibition of nitric oxide formation. Ann N Y Acad Sci. 1996;777:356–361.