
 by Kenneth Osiezagha, MD; Shahid Ali, MD; C. Freeman, MD; Narviar C. Barker, PhD; Shagufta Jabeen, MD; Sarbani Maitra, MD; Yetunde Olagbemiro, MD; William Richie, MD, FAPA; and Rahn K. Bailey, MD, FAPA
by Kenneth Osiezagha, MD; Shahid Ali, MD; C. Freeman, MD; Narviar C. Barker, PhD; Shagufta Jabeen, MD; Sarbani Maitra, MD; Yetunde Olagbemiro, MD; William Richie, MD, FAPA; and Rahn K. Bailey, MD, FAPA
All authors are from Department of Psychiatry and Behavioral Health, Meharry Medical College in Nashville, Tennessee.
Innov Clin Neurosci. 2013;10(4):26–32
Funding: No funding was provided for the preparation of this article.
Financial Disclosures: The authors do not have conflicts of interest relevant to the content of this article.
Key Words: Thiamine deficiency, Kreb’s cycle, delirium, Wernicke’s encephalopathy
Abstract: Thiamine is an essential vitamin that plays an important role in cellular production of energy from ingested food and enhances normal neuronal actives. Deficiency of this vitamin leads to a very serious clinical condition known as delirium. Studies performed in the United States and other parts of the world have established the link between thiamine deficiency and delirium. This literature review examines the physiology, pathophysiology, predisposing factors, clinical manifestations (e.g., Wernicke’s encephalopathy, Wernicke-Korsakoff syndrome, structural and functional brain injuries) and diagnosis of thiamine deficiency and delirium. Current treatment practices are also discussed that may improve patient outcome, which ultimately may result in a reduction in healthcare costs.
Introduction
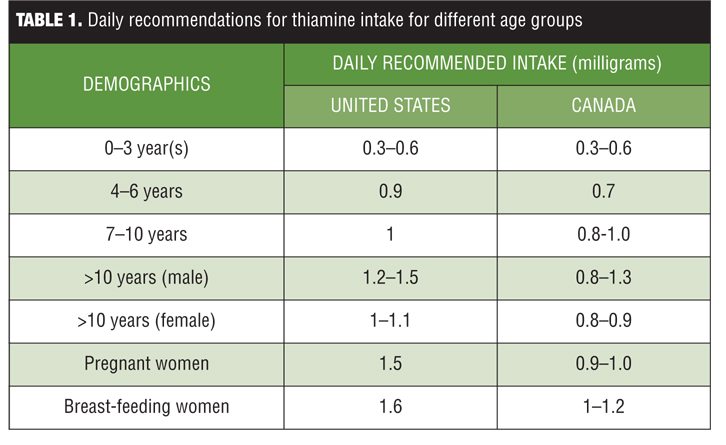
Vitamin B1, also known as thiamine, is one of the eight essential B vitamins that help the body convert food (carbohydrates, fat, and protein) into energy. These vitamins are vital for proper functioning of the central and peripheral nervous system. The human body does not produce endogenous thiamine; therefore, it must be ingested. Various dietary sources of thiamine include meat (e.g., pork, poultry), whole grain cereals (e.g., brown rice and bran), nuts, dried beans, peas, and soybeans. Breads and cereals are commonly fortified with thiamine. The human body requires a minimum of 0.33 milligrams (mg) of thiamine for every 1,000 kilocalories (kcal) it consumes, so an individual who consumes an average 2,000kcal diet per day should ingest a minimum of 0.66mg of thiamine daily.[1] A daily intake of 1.1mg of thiamine is currently recommended for adult women and 1.2mg for adult men. Children require lower levels of thiamine (0.5–1.2mg depending on age and gender), and slightly higher levels of thiamine are recommended for pregnant and breast-feeding women (1.4mg thiamine per day). Studies have found that most healthy people typically consume 0.4 to 2.0mg thiamine daily.[2] See Table 1 for a list of the daily recommended allowances for different age groups.
Physiology of Thiamine
Thiamine is a water-soluble vitamin stored primarily in the liver; however, storage only lasts up to 18 days.[3–6] The absorption of thiamine occurs in the duodenum by an active process and is converted to its active form thiamine pyrophosphate.[7,8] This conversion process of thiamine to its active form requires magnesium as a cofactor, and hence hypomagnesaemia can mimic thiamine deficiency.[6,9,10] The absorption process of thiamine is a carrier-mediated, rate-limiting process,[11] and alcohol inhibits this rate-limiting step by interfering with intestinal ATPase involved in the absorption of thiamine.[12,13] At the blood brain barrier (BBB), its transport occurs through both passive and active mechanisms[14] depending on the concentration of thiamine in the blood. When the concentration of thiamine in the blood is high, transportation of thiamine through the BBB occurs via passive diffusion. However when the concentration of thiamine in the blood is low, thiamine crosses over via active transport (Figure 1).

Pathophysiology
A deficiency in thiamine leads to decreased activity of thiamine-dependent enzymes that triggers a sequence of metabolic events leading to energy compromise. Neuronal death often occurs in certain neuronal populations that have high metabolic requirements and high thiamine turnover.[15] Areas commonly affected by thiamine deficiency are the mediodorsal thalamic nucleus, mammillary bodies, the periaqueductal gray matter, and the floor of the fourth ventricle, which includes the ocular motor, vestibular nuclei, and the cerebellar vermis.[16] Lesions also may involve the fornices, the hippocampus, the area round the third ventricle, the quadrigeminal bodies, and the cortex.[17] The predilection to affect memory circuits is responsible for the most important sequela of Wernicke’s encephalopathy— Korsakoff’s psychosis.[18]
Thiamine and enzymes in need of thiamine as a co-factor are present in all body cells. Deficiency of thiamine affects all organ systems, particularly the cells of the nervous system (e.g., neurons and glia cells, which are supporting cells in the nervous system).[19]
Thiamine pyrophosphate (TPP), the active form of thiamine, is a required cofactor in two enzyme-mediated carbohydrate metabolism pathways, which are the Kreb’s cycle (also known as the citric acid cycle) and Pentose phosphate pathway. The Kreb’s cycle is a central metabolic pathway that completes the oxidative degradation of monosaccharide (carbohydrate) and other nutrients, such as fatty acids and amino acids, and occurs in the mitochondria of every cell that utilizes oxygen. The diagram in Figure 2 shows the utilization of thiamine (TPP) in the Kreb’s cycle.

There are two main enzymes, pyruvate dehydrogenase complex (PDH) and alpha-ketoglutarate dehydrogenase (alpha-KGDH) that require thiamine as a cofactor in the Kreb’s cycle. The main function of the pathway is the generation of a molecule called adenosine triphosphate (ATP), which provides energy for numerous cellular processes and reactions.20 A decrease in ATP production in the brain leads to an increase in production of toxic metabolites of dopamine and inhibition of catechol-O-methyl transferase (COMT) activities, which is a vital enzyme for synthesis and breakdown of dopamine in the prefrontal cortex.[21,22] Thus, an increase in the level of dopamine may lead to delirium, including hallucinations and delusions.[22] In addition, proper functioning of PDH is essential for the production of the neurotransmitter acetylcholine as well as for the synthesis of myelin.20 Low levels of acetylcholine have been linked to delirium as well as the severity of cognitive impairment associated with delirium.[14,23] The citric acid cycle and alpha-KGDH play a role in maintaining the levels of the glutamate, gamma aminobutyric acid (GABA), and aspartate.[24,25] GABA is a major inhibitory neurotransmitter in the brain, and a decrease in the levels of this neurotransmitter and an increase in its amino acid precursor, glutamine, leads to neuronal excitation and hence delirium.[14]
Pentose phosphate pathway (PPP) is a very important pathway that occurs in the cytosol of human cells, including the brain, and serves as an alternative pathway to glycolysis.[26] Transketolase is an important enzyme in the pentose phosphate pathway that requires thiamine as a cofactor, modifying a molecule called glucose-6-phosphate, which is derived from glucose. This leads to the production of compounds; ribose-5-phosphate and reduced nicotinamide adenine dinucleotide phosphate (NADPH). Both of these molecules are essential for the production of numerous other important molecules in the cell. Ribose-5-phosphate is needed for the synthesis of nucleic acids, complex sugar molecules, and other compounds. NADPH provides hydrogen atoms for chemical reactions that result in the production of steroids, fatty acids, amino acids, certain neurotransmitters, and other molecules. In addition, NADPH plays an important role in the synthesis of glutathione, a compound that is essential in the body’s defense against oxidative stress. Glutathione converts reactive H2O2 into H2O. Impairment of this process leads to conversion of H2O2 to hydroxyl free radicals, which causes cellular injuries in neuronal cells, which presents as delirium.[27]
Predisposing Factors to Thiamine Deficiency
Thiamine deficiency can result from conditions that restrict eating, such as orofacial cancers, or limit adequate vitamin absorption, such as gastric bypass surgery,[28] gastric and colon cancer, hyperemesis gravidarum, or starvation by choice or associated with sepsis, surgical complications, and coma.[27,29,30] By far the most common cause of thiamine deficiency throughout the world is alcoholism,[31] as described in landmark studies of alcoholic Wernicke-Korsakoff syndrome (WKS).[32,33] Individuals with alcoholism are at special risk for thiamine deficiency because of poor diet associated with their lifestyle and because chronic alcoholism compromises thiamine absorption from the gastrointestinal tract, impairs thiamine storage, and may reduce thiamine phosphorylation essential for cellular function.[31,34–37] Table 2 outlines the predisposing factors for thiamine deficiency.

Diagnosis
Wernicke’s encephalopathy (WE) remains a clinical diagnosis; however, the best aid for a correct diagnosis is having a high index of clinical suspicion. Diagnosis includes taking a good history, looking out for causes of thiamine deficiency, and physical examination for expected signs (e.g., edema, problems with balance).
Laboratory testing, although not specific for thiamine deficiency, include measurements of some carbohydrate metabolites that accumulate in the body due to impairment of carbohydrate metabolism. These metabolites include blood levels of lactate and pyruvate.[38] Blood levels of magnesium should also be checked because magnesium is required for the conversion of thiamine into its active form, thiamine pyrophosphate.[39]
A presumptive diagnosis of WE can be confirmed by determining blood thiamine concentrations or by measuring the erythrocyte transketolase activity.[40,41] This test is limited by its lack of specificity.[42] Recently, an isocratic high performance liquid chromatography method for the assessment of thiamine, thiamine monophosphate, and thiamine diphosphate in human erythrocytes has been described.[43] This procedure has improved reproducibility, practicability, and performance compared with previous methods, and is suitable for both clinical and research purposes.
Other routine diagnostic studies include lumbar puncture, electroencephalography, and neuroimaging studies. Cerebrospinal fluid is normal in most patients, although raised protein concentrations can occur in late stages. Results of electroencephalography are within normal limits at an early stage, but show nonspecific slowing of the dominant rhythm in a late stage. Among paraclinical studies, magnetic resonance imaging (MRI) is currently considered the most valuable method to confirm a diagnosis of WE. MRI has a sensitivity of only 53 percent, but its high specificity of 93 pecent means that it can be used to rule out the disorder.[8,44]
MRI studies typically show an increased T2 signal, bilaterally symmetrical, in the paraventricular regions of the thalamus, the hypothalamus, mamillary bodies, the periaqueductal region, the floor of the fourth ventricle, and midline cerebellum.[8,45] Disruption of the BBB has been seen in these regions in 6 of the 12 patients studied with contrast-enhanced computed tomography (CT) or MRI scans.[45] Importantly, the typical pattern of lesions on an MRI is observed in only 58 percent of patients.[46] Unusual sites of lesions include cortical regions and the splenium of the corpus callosum. Valuable adjuncts for an early diagnosis of Wernicke’s encephalopathy are diffusion weighted MRI and proton MR spectroscopy,[47] which also may provide information on the pathophysiology of this encephalopathy. Ultimately, the diagnosis in a living patient is mainly supported by the response of neurological signs to parenteral thiamine.[38]
Clinically, early detection of thiamine deficiency is a difficult task because symptoms can be vague and nonspecific such as frequent headaches, fatigue, irritability, abdominal discomfort, and in children, decline in the growth rate.[6] However, thiamine deficiency presents acutely with the life-threatening condition WE. WE has an estimated mortality rate of about 20 percent.[11,48] This condition is marked by lesions of periventricular nuclei, hypothalamic nuclei, tectal plate and thalamus. Involvement of these organs resulting in WE’s cardinal signs of ophthalmoplegia, nystagmus, ataxia, and delirium with significant spatial (the inability of a person to determine his true body position, motion, and altitude relative to the earth or his surroundings) and temporal (a state in which an individual has no bearing of time) disorientation.[33,49] Infarction of the mammillothalamic tracts is another antecedent of WE. About 82 percent of patients with WE present with delirium according to autopsy-based series.[32] This constellation of symptoms is largely due to involvement of thalamic or mamillary bodies and range from a confusional state to mental sluggishness, apathy, impaired awareness of the immediate situation, inability to concentrate and, if left untreated, coma and death.[32,50] Some patients may present with confusion or agitation, hallucinations, and behavioral disturbances that mimic an acute psychotic disorder.[51,52] Ocular abnormalities occur in approximately 29 pecent of patients, and include nystagmus, symmetrical or asymmetrical palsy of either lateral recti or other ocular muscles, and conjugate-gaze palsies, which result from lesions of the pontine tegmentum and of the abducens and oculomotor nuclei.[53] Acute WE, if left untreated, leads to the more profound, debilitating, global amnesic condition of Korsakoff’s psychosis (KS),[54] although WE does not necessarily evolve into KS if adequately treated.[11,33] It remains controversial whether KS can develop without WE and whether mild forms of neuropsychological sequelae arise from repeated bouts of thiamine deficiency or inadequate treatment of such episodes.
About 80 percent of cases of WE-KS diagnosis are made during autopsy.[55] This might be caused by either a relatively nonspecific clinical presentation of the disease in some cases or poor clinical recognition of the neurological signs by clinicians. Studies have shown that the possibility of an incorrect diagnosis is high in alcohol-dependent patients presenting to accident and emergency departments.[11] The common signs of WE are difficult, if not impossible, to differentiate from intoxication with alcohol, and such problems need to be addressed with emergency medicine doctors and nurses as part of their training.[25] The lack of an adequate training and preparation may result in misdiagnosis and treatment.[29,56] There are no specific routine laboratory tests available, and no specific diagnostic abnormalities have been revealed in cerebrospinal fluid, brain imaging, electroencephalogram, or evoked potentials.
Treatment
Thiamine deficiency is the established cause of WKS. WKS usually occurs in individuals with chronic alcoholism and affects the central nervous system (brain and spinal cord). WKS is the foremost neurological complication of thiamine (vitamin B1) deficiency,[50] and the term WKS refers to two different syndromes, each representing a different stage of the disease: WE is an acute syndrome requiring emergent treatment to prevent death and neurological morbidity; and Korsakoff syndrome (KS) refers to a chronic neurological condition that usually occurs as a consequence of WE.[50,57]
The treatment of clinical manifestations of thiamine deficiency, especially WE is a medical emergency, and in patients in whom the disorder is suspected thiamine should be initiated immediately, either intravenously or intramuscularly, to ensure adequate absorption.[58] Studies[11,31,48] support several different regimens for patients with the disorder and those at risk of developing it. Specifically, patients who have signs indicative of WE should be treated empirically with a minimum of 500mg thiamine hydrochloride per 100mL of normal saline, given by infusion over a period of 30 minutes, three times per day for 2 to 3 days. In cases where there is no response, supplementation may be discontinued after 2 to 3 days. In cases where an effective response is observed, 250mg thiamine should be given intravenously or intramuscularly daily for 3 to 5 days, or until clinical improvement ceases, and should be continued. Doses of thiamine below 250mg per day apparently may not restore vitamin status,[59,60] improve clinical signs,[61] or prevent death.[62] In particular, when patients with WE are inappropriately treated with low doses of thiamine, the biochemical abnormalities caused by thiamine deficiency can lead to irreversible brain damage and death.[11,48] Recent data from a controlled study into the therapeutic benefits of thiamine in alcohol-dependent patients without clinically apparent WE indicate that at least 200mg of parenteral thiamine may be required to improve neurological symptoms.[63] Prophylactic treatment with intravenous or intramuscular administration of 100mg per day (in the United States[64]) or 250mg once daily in for 3 to 5 consecutive days should be used in all patients with severe alcohol withdrawal and poor diet with signs of malnutrition.[11,48,62] It is mandatory that thiamine is given before or concomitantly with intravenous administration of glucose so as not to precipitate WE. The changes in mental status have shown to improve after 2 to 3 weeks of therapy. Deficiency in other vitamins and electrolytes, especially niacin and magnesium, also should be corrected. Thiamine supplementation by mouth should be continued for several months at a dose of 30mg twice daily, since it is not toxic to the body in excess amounts.[59]
Typical brain lesions of WE are observed at autopsy in 0.8 to 2.8 percent of the general population in the Western world, and the vast majority of affected patients are alcoholic.[50,65] The prevalence of WE lesions seen on autopsy in alcohol abusers is generally higher than in those individuals who do not abuse alcohol.[66] Among those with alcohol-related deaths, it has been reported to be even higher: 29 to 59 percent.[67,68] Cases of WE in men generally outnumber cases in women; yet women appear more susceptible to developing WE than men.[50,69]
Conclusion
Thiamine is an essential requirement for normal cerebral energy metabolism. Deficiency of thiamine leads to mitochondria dysfunction, which manifests as WE/KS. The underlying pathophysiology of thiamine deficiency is multifactorial in nature and involves a wide-ranging cascade of events that involves impairment of glucose metabolism, ultimately resulting in neuronal cell death. Additional studies are required to explain the predilection of selective brain structures due to thiamine deficiency. Also, randomized, controlled trials are needed to define the optimum dose and duration of thiamine treatment for prophylaxis or treatment of clinical manifestations of thiamine deficiency. Clinicians and other ancillary health workers should be properly trained to have a high index of suspicion of thiamine deficiency in situations of unbalanced nutrition that has lasted for 2 to 3 weeks to avoid misdiagnosis of the life threatening WE or irreversible brain damage caused by KS. Finally, while thiamine deficiency involves a variety of functional impairments at several different levels, understanding the mechanisms that underlie the characteristic focal vulnerability associated with this disorder remains the primary goal and is expected to continue to yield important new insight into how derangement in the metabolic process leads to life-threatening neuropsychiatric conditions.
References
1. Hoyumpa AM Jr. Mechanisms of thiamin deficiency in chronic alcoholism. Am J Clin Nutr. 1980;33(12):2750–2761.
2. Wöhrle JC, Spengos K, Steinke W, et al. Alcohol-related acute axonal polyneuropathy: a differential diagnosis of Guillain-Barre syndrome. Arch Neurol. 1998;55(10):1329–1334.
3. Netravathi M, Sinha S, Taly AB, et al. Hyperemesis gravidarum-induced Wernicke’s encephalopathy. J R Coll Physicians Edinb. 2009;39:125–128.
4. Isselbacher KJ, Braunwald E, Wilson JD. Harrison’s Principles of Internal Medicine, Thirteeth Edition. New York: McGraw-Hill; 1994.
5. McCormick D. Modern Nutrition in Health and Disease. Philadelphia: Lea and Febiger; 1988.
6. Shikata E, Mizutani T, Kokubun Y, Takasu T. “Iatrogenic” Wernicke’s encephalopathy in Japan. Eur Neurol,. 2000;44(3):156–161.
7. Mahan LK, Escott-Stump S. Krause’s Food, Nutrition, and Diet Therapy, Tenth Edition. Philadelphia: W.B. Saunders Company; 2000.
8. Antunez E, Estruch R, Cardenal C, et al. Usefulness of CT and MR imaging in the diagnosis of acute Wernicke’s encephalopathy. Am J Roentgenol. 1998;171(4):1131–1137.
9. Sechi G, Serra A. Wernicke’s encephalopathy: new clinical settings and recent advances in diagnosis and management. Lancet Neurol.2007;6(5):442–455.
10. Zieve L. Influence of magnesium deficiency on the utilization of thiamine. Ann N Y Acad Sci. 1969;162(2):732–743.
11. Tallaksen CM, Bell H, Bohmer T. Thiamin and thiamin phosphate ester deficiency assessed by high performance liquid chromatography in four clinical cases of Wernicke encephalopathy. Alcohol Clin Exp Res. 1993;17(3):712–716.
12. Coombs G. The Vitamins: Fundamental Roles in Nutrition and Health. New York: Academic Press; 1992.
13. Todd KG, Hazell AS, Butterworth RF. Alcohol-thiamine interactions: an update on the pathogenesis of Wernicke encephalopathy. Addict Biol. 1999;4(3):261–272.
14. Thomson AD. Mechanisms of vitamin deficiency in chronic alcohol misusers and the development of the Wernicke-Korsakoff syndrome. Alcohol Alcohol. 2000;35(Suppl 1):2–7.
15. Hazell AS, Todd KG, Butterworth RF. Mechanisms of neuronal cell death in Wernicke’s encephalopathy. Metabolic Brain Disease. 1998;13:97–122.
16. Baker KG, Harding AJ, Halliday GM, et al. Neuronal loss in functional zones of the cerebellum of chronic alcoholics with and without Wernicke’s encephalopathy. Neuroscience. 1999;91(2):429–438.
17. Torvik A. Brain lesions in alcoholics: neuropathological observations. Acta Med Scand. 1987;717(Suppl):47–54.
18. Kessels RPC, Kortrijk HE, Wester AJ, Nys GMS. Confabulation behavior and false memories in Korsakoff’s syndrome: role of source memory and executive functioning. Psychiatry Clin Neurosci. 2008;62: 220–225.
19. Hoffman-LaRoche F. Vitamins (Basics). New York: Seaboard Lithographers; 1994.
20. Butterworth RF. Thiamin. In: Shils ME (ed). Modern Nutrition in Health and Disease, Tenth Edition. Baltimore, MD: Lippincott Williams & Wilkins; 2006.
21. Gunther ML, Morandi A, Ely EW. Pathophysiology of delirium in the intensive care unit. Crit Care Clin. 2008;24(1):45-65, viii.
22. Maldonado JR. Pathoetiological model of delirium: a comprehensive understanding of the neurobiology of delirium and an evidence-based approach to prevention and treatment. Crit Care Clin. 2008;24(4):789–856, ix.
23. Gibson GE, Jope R, Blass JP. Decreased synthesis of acetylcholine accompanying impaired oxidation of pyruvic acid in rat brain minces. Biochem J. 1975;148(1):17–23.
24. Purves D. Neuroscience. Sunderland, MA: Sinauer Associates, Inc.; 2008.
25. Bak LK, Schousboe A, Waagepetersen HS. The glutamate/GABA-glutamine cycle: aspects of transport, neurotransmitter homeostasis and ammonia transfer. J Neurochem. 2006.98(3):641–653.
26. Kruger NJ, von Schaewen A. The oxidative pentose phosphate pathway: structure and organisation. Curr Opin Plant Biol. 2003;6(3):236–246.
27. Martin PR, Adinoff B, Weingartner H, et al. Alcoholic organic brain disease: nosology and pathophysiologic mechanisms. Prog Neuropsychopharmacol Biol Psychiatry. 1986;10(2):147–164.
28. Worden RW, Allen HM. Wernicke’s encephalopathy after gastric bypass that masqueraded as acute psychosis: a case report. Curr Surg. 2006;63(2):114–116.
29. Thomson AD, Cook CC, Touquet R, et al. The Royal College of Physicians report on alcohol: guidelines for managing Wernicke’s encephalopathy in the accident and emergency department. Alcohol Alcohol. 2002;37(6):513–521.
30. Sechi G, Serra A, Pirastru MI, et al. Wernicke’s encephalopathy in a woman on slimming diet. Neurology. 2002;58(11):1697–1698.
31. Thomson AD, Marshall EJ. The treatment of patients at risk of developing Wernicke’s encephalopathy in the community. Alcohol Alcohol. 2006;41(2):159–167.
32. Harper CG, Giles M, Finlay-Jones R. Clinical signs in the Wernicke-Korsakoff complex: a retrospective analysis of 131 cases diagnosed at necropsy. J Neurol Neurosurg Psychiatry. 1986;49(4):341–345.
33. Tune LE, Damlouji NF, Holland A, et al. Association of postoperative delirium with raised serum levels of anticholinergic drugs. Lancet. 1981;2(8248):651–653.
34. Thomson AD, Marshall EJ. The natural history and pathophysiology of Wernicke’s Encephalopathy and Korsakoff’s Psychosis. Alcohol Alcohol. 2006;41(2):151–158.
35. Lieber CS. Relationships between nutrition, alcohol use, and liver disease. Alcohol Res Health. 2003;27(3):220–231.
36. Martin PR, Singleton CK, Hiller-Sturmhöfel S. The role of thiamine deficiency in alcoholic brain disease. Alcohol Res Health. 2003;27(2):134–142.
37. Leevy CM. Thiamin deficiency and alcoholism. Ann N Y Acad Sci. 1982;378:316–326.
38. Chamberlin SL, Narins B. The Gale Encyclopedia of Neurological Disorders. Detroit, MI: Thomson Gale: Detroit; 2005.
39. Woodhill JM, Nobile S. Thiamine in the 1970 Australian diet with special reference to cereals and the assessment of thiamine status. Int J Vitam Nutr Res. 1972;42(3):435–443.
40. Dreyfus PM. Clinical application of blood transketolase determinations. N Engl J Med. 1962;267:596–598.
41. Baker H, Frank O, Fennelly JJ, Leevy CM. A method for assaying thiamine status in man and animals. Am J Clin Nutr. 1964;14:197–201.
42. Victor M, Adams RD, Collins GH. The Wernicke-Korsakoff syndrome. a clinical and pathological study of 245 patients, 82 with post-mortem examinations. Contemp Neurol Ser. 1971;7:1–206.
43. Mancinelli R, Ceccanti M, Guiducci MS, et al. Simultaneous liquid chromatographic assessment of thiamine, thiamine monophosphate and thiamine diphosphate in human erythrocytes: a study on alcoholics. J Chromatogr B Analyt Technol Biomed Life Sci. 2003;789(2):355–363.
44. Chung SP, Kim SW, Yoo IS, et al. Magnetic resonance imaging as a diagnostic adjunct to Wernicke encephalopathy in the ED. Am J Emerg Med. 2003;21(6):497–502.
45. Mascalchi M, Simonelli P, Tessa C, et al. Do acute lesions of Wernicke’s encephalopathy show contrast enhancement? Report of three cases and review of the literature. Neuroradiology. 1999;41(4):249–254.
46. Victor M. The Wernicke-Korsakoff syndrome. In: Vinken PJ, Bruyn GW (eds). Handbook of Clinical Neurology. Amsterdam: North-Holland Publishing Company; 1976.
47. Rugilo CA, Uribe Roca MC, et al. Proton MR spectroscopy in Wernicke encephalopathy. AJNR Am J Neuroradiol. 2003;24(5):952–955.
48. Cook CC, Hallwood PM, Thomson AD. B Vitamin deficiency and neuropsychiatric syndromes in alcohol misuse. Alcohol Alcohol. 1998;33(4):317–336.
49. Yoneoka Y, Takeda N, Inoue A, et al. Acute Korsakoff syndrome following mammillothalamic tract infarction. AJNR Am J Neuroradiol. 2004;25(6):964–968.
50. Victor M, Adams RD, Collins GH. The Wernicke-Korsakoff Syndrome (WKS) and Related Neurological Disorders Due to Alcoholism and Malnutrition, Second Edition. Philadelphia: FA Davie; 1989.
51. Jiang W, Gagliardi JP, Raj YP, et al. Acute psychotic disorder after gastric bypass surgery: differential diagnosis and treatment. Am J Psychiatry. 2006;163(1):15–19.
52. Wooley JA. Characteristics of thiamin and its relevance to the management of heart failure. Nutr Clin Pract. 2008;23(5):487–493.
53. van der Mast RC. Pathophysiology of delirium. J Geriatr Psychiatry Neurol. 1998;11(3):138–145; discussion 157–158.
54. Zubaran C, Fernandes JG, Rodnight R. Wernicke-Korsakoff syndrome. Postgrad Med J. 1997;73(855):27–31.
55. Thomson AD, Guerrini I, Marshall EJ. Wernicke’s encephalopathy: role of thiamine. Pract Gastroenterol. 2009;33(6):21–30.
56. Agabio R. Thiamine administration in alcohol-dependent patients. Alcohol Alcohol. 2005;40(2):155–156.
57. Charness ME, So YT Wernicke’s encephalopathy. Up to Date. Wolters Kluwer Health. http://www.uptodate.com/contents/wernickes-encephalopathy?source=search_result&search=Charness+ME%2C+So+YT+Wernicke%27s+encephalopathy.&selectedTitle=1%7E150. Access date April 1, 2013.
58. Tanphaichitr V. Thiamin. In: Shils ME (ed). Modern Nutrition in Health and Disease, Ninth Edition.Baltimore, MD: Williams and Wilkins; 1999.
59. Hope LC, Cook CC, Thomson AD. A survey of the current clinical practice of psychiatrists and accident and emergency specialists in the United Kingdom concerning vitamin supplementation for chronic alcohol misusers. Alcohol Alcohol. 1999;34(6):862–867.
60. Brown LM, Rowe AE, Ryle PR, et al. Efficacy of vitamin supplementation in chronic alcoholics undergoing detoxification. Alcohol Alcohol. 1983;18(2):157–166.
61. Schenker S, Henderson GI, Hoyumpa AM Jr, et al. Hepatic and Wernicke’s encephalopathies: current concepts of pathogenesis. Am J Clin Nutr. 1980;33(12):2719–2726.
62. Cook CC. Prevention and treatment of Wernicke-Korsakoff syndrome. Alcohol Alcohol Suppl. 2000;35(1):19–20.
63. Ambrose ML, Bowden SC, Whelan G. Thiamin treatment and working memory function of alcohol-dependent people: preliminary findings. Alcohol Clin Exp Res. 2001;25(1):112–116.
64. Vitamin B1 (thiamine). Up to Date. Wolters Kluwer Health. http://www.uptodate.com/contents/search?search=Vitamin+B1+%28thiamine%29&x=0&y=0. Accessed April 1, 2013.
65. Torvik A. Wernicke’s encephalopathy—prevalence and clinical spectrum. Alcohol Alcohol Suppl. 1991;1:381–384.
66. Charness ME, Simon RP, Greenberg DA. Ethanol and the nervous system. N Engl J Med. 1989;321(7):442–454.
67. Torvik A, Lindboe CF, Rogde S. Brain lesions in alcoholics. A neuropathological study with clinical correlations. J Neurol Sci. 1982;56(2-3):233–248.
68. Naidoo DP, Bramdev A, Cooper K. Autopsy prevalence of Wernicke’s encephalopathy in alcohol-related disease. S Afr Med J. 1996;86(9):1110–1112.
69. Skullerud K, Andersen SN, Lundevall J. Cerebral lesions and causes of death in male alcoholics. a forensic autopsy study. Int J Legal Med. 1991;104(4):209–213.