

by William P. Horan, PhD; Gary Sachs, MD; Dawn I. Velligan, PhD; Michael Davis, MD, PhD; Richard S.E. Keefe, PhD; Ni A. Khin, MD; Florence Butlen-Ducuing, MD, PhD; and Philip D. Harvey, PhD
Dr. Horan is with Karuna Therapeutics in Boston, Massachusetts, and University of California in Los Angeles, California. Dr. Sachs is with Signant Health in Boston, Massachusetts, and Harvard Medical School in Boston, Massachusetts. Dr. Velligan is with University of Texas Health Science Center at San Antonio in San Antonio, Texas. Dr. Davis is with Usona Institute in Madison, Wisconsin. Dr. Keefe is with Duke University Medical Center in Durham, North Carolina. Dr. Khin is with Neurocrine Biosciences, Inc. in San Diego, California. Dr. Butlen-Ducuing is with Office for Neurological and Psychiatric Disorders, European Medicines Agency in Amsterdam, The Netherlands. Dr. Harvey is with University of Miami Miller School of Medicine in Miami, Florida.
Funding: No funding was provided for this article.
Disclosures: WPH is an employee of and holds equity in Karuna Therapeutics. GS is an employee of Signant Health and is a consultant for Merck, Intra-Cellular, and Biohaven. DIV has been an Advisory Board speaker for Teva and Otsuka, and an Advisory Board Participant for Janssen, Indivior, and Boehringer Ingelheim. MD is an employee of the Osuna Institute. RSEK serves as a consultant to WCG, Karuna, Novartis, Kynexis, Gedeon-Richter, Pangea, Merck, and Boehringer-Ingelheim, and receives royalties for the BAC, BACS and VRFCAT. NAK is an employee of Neurocrine Biosciences, Inc. FB-D: The views expressed in this article are the personal views of the authors and may not be understood or quoted as being made on behalf of or reflecting the position of the European Medicines Agency or one of its committees or working parties. PDH has received consulting fees or travel reimbursements from Alkermes, Boehringer Ingelheim, Karuna Therapeutics, Merck Pharma, Minerva Neurosciences, and Sunovion (DSP) Pharma in the past year; receives royalties from the Brief Assessment of Cognition in Schizophrenia (Owned by WCG Endpoint Solutions, Inc. and contained in the MCCB); and is chief scientific officer of i-Function, Inc and scientific consultant to EMA Wellness, Inc.
Innov Clin Neurosci. 2024;21(1–3):19–30.
Abstract
Excessive placebo response rates have long been a major challenge for central nervous system (CNS) drug discovery. As CNS trials progressively shift toward digitalization, decentralization, and novel remote assessment approaches, questions are emerging about whether innovative technologies can help mitigate the placebo response. This article begins with a conceptual framework for understanding placebo response. We then critically evaluate the potential of a range of innovative technologies and associated research designs that might help mitigate the placebo response and enhance detection of treatment signals. These include technologies developed to directly address placebo response; technology-based approaches focused on recruitment, retention, and data collection with potential relevance to placebo response; and novel remote digital phenotyping technologies. Finally, we describe key scientific and regulatory considerations when evaluating and selecting innovative strategies to mitigate placebo response. While a range of technological innovations shows potential for helping to address the placebo response in CNS trials, much work remains to carefully evaluate their risks and benefits.
Keywords: Placebo response, clinical trials, technology, remote assessment, ecological momentary assessment, regulatory
Placebo response rates are particularly large in central nervous system (CNS) trials1–4 and have steadily increased over the past several decades.3,5,6 The risk that placebo response will obscure signal detection has led many pharmaceutical companies to reduce or close their CNS research and development programs.7–9 The recognition of placebo response as an obstacle to CNS drug development has inspired development of innovative placebo response mitigation technology (PRMT). Putative PRMTs are intended to reduce the risk of a failed clinical trial and target nearly all phases of CNS trials (e.g., recruitment and retention, outcome assessment, data collection, and analytics). Since PRMTs are at different stages of development and uptake in CNS trials, drug developers face an overwhelming array of choices when considering new PRMTs. This manuscript is intended to aid CNS drug developers, regulators, and other stakeholders wanting to understand and critically evaluate various PRMTs.
Placebo mitigation efforts have largely focused on CNS trial design features, such as blinded placebo lead-in periods, to identify early placebo responders and then adjust participation accordingly. Unfortunately, the evidence indicates that these “blind and refine” approaches show limited success in CNS trials.10–12 Furthermore, although brief, generic placebo response education is often provided to raters at investigator meetings, there is little evidence demonstrating the impact of such training on subsequent signal detection. The evolution of CNS trials toward increased implementation of digitalization, decentralization, and remote assessment13,14 creates opportunities for innovative solutions, which have been marketed as strategies to mitigate placebo response, as well as enhance signal detection and efficiency of trial conduct. The current review provides an overview of the status and level of evidence supporting these diverse approaches.
To aid CNS drug developers and regulators seeking a starting point as they evaluate PRMTs, the manuscript is organized into three sections. First, we briefly lay out a conceptual framework defining components of placebo response. Second, we offer some critical evaluation of the promise of a range of currently available and emerging technologies in terms of their potential impact on components of placebo response. Third, we conclude by considering potential scientific and regulatory issues confronting drug developers as they evaluate and/or attempt to integrate PRMT into trials.
Conceptual Overview of the Placebo Response: A Multifaceted Phenomenon
The concept of placebo response is broad and heterogeneous.15–17 Out of necessity, clinical trials use a global operational definition in which placebo response for any primary outcome measure is simply the difference between the scores at baseline and endpoint for participants assigned to the placebo group. This difference score indexes improvement or worsening in response to an intervention with no direct physiological activity, generating a value that can be a benchmark for computing comparative efficacy. Since these change scores can reflect several distinct processes, the size of a change score alone does not provide guidance for efforts to mitigate placebo response. Therefore, we subdivide the complex topic of placebo response into more granular components. A useful initial heuristic segments operationally defined placebo response into two broad conceptual categories, which are depicted in Figure 1.

Category 1 placebo responses are those in which the patient’s target condition (e.g., depression, anxiety, psychosis) improves, and this improvement is reflected in changes in the primary outcome measure. These changes include what we define as true placebo response—reactive responses to outcome assessments or other research procedures, as well as nonstudy interventions. Category 1 responses can be largely understood as arising when expectations or experiences trigger physiological changes that result in actual improvement in the targeted condition (Table 1). These influences include patient factors and interactions between subjects and study staff. Studies by Kaptchuk et al18 and Czerniak et al19 demonstrated the clinically beneficial effect of warmly engaged interaction with study staff. Expectation effects are also powerful, in that telling subjects that placebo can be helpful for their condition has been shown to increase response even when the subject knows that the placebo is an inactive substance.20
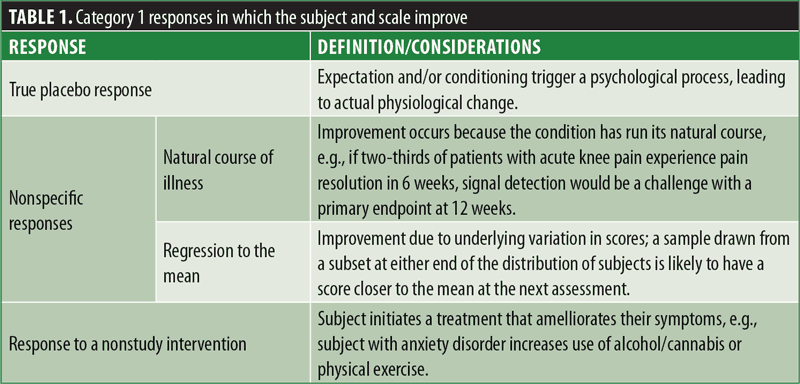
Category 2 placebo responses are those in which scores on the outcome measure change, but the patient’s target condition does not. These include instances when changes in outcome measures are attributable to measurement error, response biases, and/or misconduct on the part of raters or participants. This type of placebo response might reflect the inclusion of patients who do not truly meet entry criteria (e.g., illness or severity) due to errors in assessments, operational problems (including partial adherence to assessment outcomes), or other behaviors misaligned with the study objective (e.g., a participant or site rater exaggerating the severity of symptoms to gain entry into a clinical trial; Table 2).
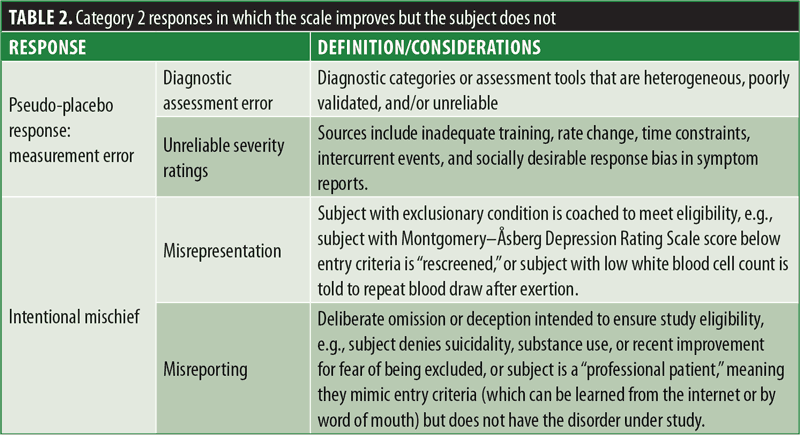
It is important to keep in mind that regardless of whether the origin of the placebo response is Category 1 or 2, a failed trial is defined by changes in the primary outcome that do not differ between active treatment and placebo. Thus, both types of placebo response require mitigation. Although Category 1 responses imply meaningful clinical change with placebo treatment, this change is detrimental to the goals of identifying treatment effects in clinical trials and advancing the approval of new treatments.
Technologies Developed to Directly Address Placebo Response
In the past decade, several PRMTs have emerged in the CNS trial area. In this section, we review four approaches: artificial intelligence (AI), genomic biomarkers, participant training, and remote assessment.
AI approaches to identifying placebo responder status. Although efforts to identify a personality trait or characteristic that predicts placebo responder status have had limited success,21,22 recent applications of sophisticated multivariate data analytic approaches seem promising. AI approaches, including machine learning (ML)-based algorithms, offer the potential to analyze large volumes of integrated data and can reveal unanticipated, complex, multivariate models of placebo response. While numerous specific ML algorithms are available, all of them generate predictive models which emerge from the interaction between the algorithm and dataset, rather than being specified a priori and tested. Predictive ML models are initially created in a training dataset and then cross-validated with independent test data, which can be continuously refined as new data is added. How this would be applied to an ongoing clinical trial, particularly of a truly novel treatment, is a challenging question.
To date, only a handful of studies have applied even retrospective ML approaches to CNS trial data, with these studies examining data collected at or before baseline during previously completed randomized, controlled trials (RCTs). To illustrate these approaches, three studies in major depressive disorder (MDD) examined: 1) 11 demographic, clinical, and cognitive variables in 174 older adults;23 2) 283 clinical, behavioral, neuroimaging, and electrophysiological variables 141 adults;24 and 3) 18 demographic, clinical, and cognitive variables in 112 adolescents.25 All three studies reported statistically significant predictive models based on a subset of the variables examined in training datasets and confirmed the models in test datasets. In addition, two of the studies developed subject-level calculators to predict placebo response, which showed moderate predictive accuracies. A few other studies using ML approaches found that variables such as self-reported treatment expectancies, sudden atypically large improvements on an outcome measure, and resting state functional magnetic resonance imaging (fMRI) functional connectivity characteristics significantly predicted elevated placebo response in studies of depression and pain.26–29
Currently, promising proof-of-concept AI studies are emerging.30 No CNS RCT to date has used an AI-based placebo response calculator to manage enrollment; prospective studies that implement this approach are needed. There are also some important issues to consider regarding the acceptability of this approach. For example, ML-based models can be complex and nonintuitive, making it challenging for stakeholders (e.g., regulators or investors) to understand exactly how the algorithms work; this is a particular concern for algorithms that are not fixed. Furthermore, the implications of restricting enrollment based on potentially inaccurate models raises pragmatic, ethical, and regulatory concerns (discussed below).
Genomic biomarkers of placebo response. Advances in neuroimaging and genotyping technologies support the view that a true placebo response reflects a biological response triggered by cues that accompany the administration of inactive treatments.31–33 For example, experimental neuroscience studies in pain, Parkinson’s disease, and depression indicate that certain neurotransmitters, including endogenous opioids, dopamine, endocannabinoids, and serotonin, are more strongly activated in individuals showing a larger placebo response.34–38 Such findings have inspired efforts to identify candidate genes linked to the synthesis, signaling, and metabolism of these transmitters that relate to placebo responder status. Although the study of genomic effects on the placebo response, or the placebome, is very early in its development, there have been a number of intriguing findings from retrospective studies. Several single nucleotide polymorphisms (SNPs) related to functional characteristics of the neurotransmitters systems noted above (e.g., catechol-O-Methyltransferase [COMT], mu-opioid receptor gene [OPRM1]) have shown replicable associations with placebo response.33
Further investigation of genetic factors holds the promise of identifying an objective, stable trait or combination of traits to prospectively identify subjects with low potential for placebo response for sample enrichment. However, considerable work remains to translate the emerging, complex placebome into strategies that can be applied to CNS trials. For example, we do not yet know whether the sensitivity of available assays to detect placebo responder status is adequate for trial use; we do know that genomic contributions measured by genome-wide association studies (GWAS) account for less than 20 percent of the variance in susceptibility to complex traits, such as depression and schizophrenia, which would not provide acceptable sensitivity for screening. In addition, clinical trial outcomes might be differentially modified by putative placebo genotypes in the placebo versus drug treatment arms (genotype x treatment interactions).32,33 This complexity is compounded when one considers the possibility of disease-specific (genotype x treatment condition x disease) or epigenetic effects on these interactions.
Placebo response mitigation training for study participants. Although CNS trials often include brief, generic education for raters at the launch of a trial, we still lack evidence that such one-time training impacts placebo response or signal detection. New patient-focused training approaches have been developed, which aim to help participants more accurately report symptoms, provide valid information about placebo-controlled trials, and moderate participant expectations. These approaches have mainly been developed and tested in the areas of pain and psychiatry.
In the area of pain, high variability in pain reporting (presumably driven at least in part by error variance or personality factors) is associated with larger placebo responses.39–43 Accurate pain reporting training (APRT) is based on the theory that patients who can accurately utilize internal cues to report pain levels are less likely to be influenced by external cues that can increase placebo response. In a series of training sessions, patients are exposed to painful stimuli at different intensities, provide pain ratings, and receive standardized feedback about the accuracy of their pain ratings (relative to the objective stimulus intensities).39 In a RCT for painful diabetic neuropathy (N=51), those who received APRT showed a significantly smaller placebo response than those who did not receive APRT (though there was no separation between active treatment vs. placebo in the trial).43 Furthermore, the placebo response rate in a RCT of neublastin for painful lumbosacral radiculopathy (LSR) that used APRT (19.1%)44 was significantly smaller in comparison to similar trials that did not use APRT (38.0%).39 These findings provide initial support for this approach to increasing the accuracy of pain reporting.
In the psychiatric area, the Placebo-Control Reminder Script (PCRS),45 an interactive procedure designed to educate patients on factors known to amplify placebo response, has been developed and tested. A rater reads a brief script about expectation biases related to treatment benefit, the purpose of a placebo-controlled trial, how interactions with clinical trial staff differ from therapeutic interactions, and the importance of attending to these biases when reporting on their symptoms. The rater then collaboratively queries the patient about their understanding of the material. The PCRS is administered at the initial study visit and each subsequent key study visit. Although the PCRS has not yet been used in a prospective, randomized trial, in a quasi-experimental study, currently depressed patients (N=137) with major depression or schizophrenia were informed that they would be randomized to receive an antidepressant or placebo. All participants actually received placebo and were randomized to PCRS and no PCRS groups. The PCRS group reported a significantly smaller placebo response (i.e., smaller reduction in depression) and significantly fewer adverse events (nocebo effect) than the no PCRS group.
Overall, initial evidence from studies in pain and psychiatry indicates that providing brief training or psychoeducation to participants, either to enhance symptom reporting accuracy or attend to placebo response-related factors, can help mitigate the placebo response. It remains to be determined whether such approaches could reduce placebo response alone versus response to both placebo and active treatment.
Remote rater-based clinical assessments. Recent years have witnessed a rapidly growing interest in shifting from conventional in-person assessments to assessments that are conducted remotely by off-site raters. This approach has already been extensively used for clinician-reported outcome (ClinRO) measures across many CNS indications. During the COVID-19 pandemic, the United States Food and Drug Administration (FDA) also signaled openness to remote collection of patient-reported outcomes (PROs) and more complex performance outcome assessments.46 For ClinROs, video or teleconferencing is used to conduct live interviews between a patient at a trial site and a remote rater (who can be blinded to study visit as well as treatment condition) from a limited pool of highly trained, tightly calibrated interviewers who can presumably achieve higher reliability than a larger group of dispersed raters. Remote clinical assessments have the potential to impact placebo response in several ways, including reducing measurement error and biases (Category 2), but they might also have an impact on Category 1 placebo responses by reducing placebo-augmenting staff-subject interactions.
To date, there are three published studies comparing remote and in-person rating assessments.47–49 One consistent finding is that remote raters blinded to study entry criteria reported lower symptom severity ratings than site-based raters at screening and baseline visits, suggesting the potential for remote raters to mitigate baseline inflation bias. Alternatively, maintaining site unawareness of entry criteria could be easily achieved with technology-based assessments where the only feedback that the site receives is whether the participant is eligible. Category 2 placebo responses can originate from the “first honest assessment” challenge, which can be bypassed when the sites are not tasked with making the determination that patients manifest a certain level of severity of impairment and hence can be randomized.
Regarding benefits for the placebo response and treatment-signal detection, the results have been mixed. In one MDD trial,47 within the placebo group, interviewer-rated improvement in depression was significantly smaller for remote assessments (conducted by video conference at 3 of the study visits), compared to site-based assessments (conducted at 5 study visits). However, this difference in placebo response was not significant when the analysis was limited to subjects who met inclusion criteria based on the remote assessments. Drug versus placebo group comparisons in the extent of response were not evaluated. In a second MDD trial,48 the placebo response did not differ between remote (video conference) and site-based assessments (conducted at all study visits for both). Notably, drug versus placebo group differences were found for site-based ratings, but not for remote ratings. Finally, in a generalized anxiety disorder trial,49 within the placebo group, the placebo response was significantly smaller for remote assessments (conducted by telephone at 2 study visits), compared to site-based assessments (conducted at 6 study visits). Drug versus placebo group differences were not evaluated.
In this context, it is worth noting the findings from the large Phase III Rapastinel program for treatment-resistant depression,50 which found no difference in the placebo response rates or drug versus placebo separation across three essentially identical trials, including two trials with remote ratings and one trial with site-based ratings. In contrast, longitudinal information across trials supported the use of remote assessments in the valbenazine development program for tardive dyskinesia. The company’s earlier studies using site-based raters showed considerable placebo response, but their pivotal studies used central raters who were unaware of the study visit sequence. The ratings were based on recorded video rather than live interviews, so it was easier to maintain rater blindness to visit sequence. This approach appeared to markedly reduce the placebo response and suggests that rater bias based on knowledge of the time course of treatment to date can impact on their ratings.51
In summary, there is no clear signal that remote clinical assessments facilitate separation of drug and placebo arms. Within studies of the same treatment, evidence is also mixed, suggesting that remote ratings do not provide a guarantee of reduced placebo effects.
Trial Technologies with Potential Relevance to Placebo Response
In this section, we consider various potential trial technologies that could largely impact Category 2 placebo response related to the recruitment and retention, data collection, and signal analytic components of a trial.
Recruitment, exposure to assessments, and retention. While greater public awareness can positively affect drug development, widespread media coverage of clinical trials and novel drugs impact how people view clinical trials in ways that can increase placebo effects.52 For example, the large-scale media coverage surrounding dramatic improvements seen in early trials of putative, fast-acting antidepressants, such as ketamine or psilocybin, can engender widespread expectations in those joining a trial.53,54 Moreover, increased information might attract more individuals who are not appropriate for a trial who then exaggerate symptoms to be included. While studies show that using social media can increase recruitment into trials,55 there has been little discussion of potential drawbacks.
With respect to recruitment and retention, electronic informed consent (e-consent) using a variety of digital tools has been increasingly studied as an alternative to an in-person paper review consent process.56 Use of a single common presentation describing study procedures, risks, and benefits during the consent process might keep participant expectations more consistent across sites and control investigator/participant interactions that can increase placebo response.57 Literature reviews suggest that e-consent can increase the knowledge and understanding of important clinical trial concepts, such as placebo and blinding, but there is no published evidence for an impact of e-consent on placebo response rates or signal detection in an actual clinical trial.56,57
Data collection methodology. Repeating an assessment, also known as tandem ratings, offers a simple solution to the common problem of unreliable reporting prior to randomization. Max Hamilton advocated the use of two raters to administer the Hamilton Depression Rating Scale (HAM-D; 1960) and recommended declaring highly discordant ratings to be invalid rather than attempting to resolve substantial differences between the raters.58 Several global clinical trials have adopted variations on this technique for excluding subjects with highly discordant ratings. Pre-planned (mania trial)59 and post hoc analyses (multiple depression trials)60 revealed better signal detection in subjects whose baseline diagnosis or severity ratings were concordant between a site-based rater and a central rater or computer-simulated rater (CSR). Post hoc analyses show that limiting enrollment to subjects for whom tandem Montgomery–Åsberg Depression Rating Scale (MADRS) ratings were concordant prior to randomization is associated with better signal detection in successful, as well as negative, trials.59–61 There are no prospective trials reporting results for subject selection based on tandem rating criteria versus ratings without exclusion.
A variety of digital and electronic tools exist to standardize and improve data collection processes during clinical trials, and these can impact placebo response. Electronic clinical outcome assessments (eCOAs) use digital platforms to collect patient-reported, observer-reported, and clinician-rated data. Conventional pen-and-paper COAs, including performance-based outcomes, are increasingly being transformed into digital formats for CNS trials. Digital platforms that guide raters through clinical interviews in a systemic manner provide an opportunity to decrease variability in administration, reduce informal interpersonal exchanges during data collection,62,63 and alert the interviewer to out-of-range entries or potential inconsistencies in ratings. Some eCOA systems also provide the opportunity to record audio and/or video of interviewers, which can be reviewed and used to provide feedback to raters about interview quality; additionally, feedback specifically designed to keep placebo-augmenting effects to a minimum can be provided to raters.
Digital or video adherence monitoring (e.g., capturing video as participants take their medication using a smartphone application [app]) targets measurement error by identifying individuals who are not taking study medication during screening and has the added benefit of prompting adherence during a trial, which is likely to increase placebo versus treatment differences if a drug is efficacious.64 Data illustrate that virtual digital observation technology or the use of electronic observational devices can improve adherence, but data have not addressed placebo response rates.65
CSRs have been used in mood disorder studies for nearly 20 years. A CSR is a computer program that conducts an interview and determines a score on standard ClinROs. The CSRs present questions in the form of text or voice and ask subjects to select their response from a range of multiple-choice options. The CSR then uses an algorithm to select the next question and map the responses onto an anchor point for each item. This technique has been used for tandem ratings in global trials as a quality assurance technique, but there are no data on the impact for placebo response.66
An adjacent theoretical possibility involves using virtual reality (VR) technology to administer clinical rating scales in clinical trials using an avatar.67 This type of approach could directly control human factors and standardize “interviewer” (chatbot) behavior across trial visits and trial sites. Interactive VR environments and voice recognition systems are already increasingly common in assessment and intervention contexts, both office- and home-based.68–72
Prospective blinded data analytics. The adoption of eCOA systems, which capture clinical trial data in real-time, creates opportunities to apply blinded data analytics throughout the course of a clinical trial. These analytics are currently used to identify subjects, raters, or sites with unusual response profiles (e.g., extreme variation or unexpected dramatic improvements between visits, identical ratings across visits) for further evaluation during the course of a clinical trial. Actionable findings can improve signal detection by remediating problems or ending enrollment at sites with high rates of error. One goal of blinded data analytics is to detect patterns in clinical trial data that are associated with elevated placebo response and reduced drug-placebo separation. PRMTs of this type use quality metrics and statistical tests to quickly identify and potentially remediate measurement/rating errors that contribute to inflated placebo response. Several recent retrospective studies have conducted post hoc analyses of CNS trial data to identify relevant response patterns, finding that unusual or erratic ratings were associated with inflated placebo response at the subject and site levels.73–76
Emerging Data Collection Approaches: Digital Phenotyping
A wide array of mobile digital devices that people carry (e.g., smartphones) or wear (e.g., on the wrist, insoles) are increasingly used for clinical trial data acquisition. These novel digital phenotyping (DP) methods have the potential to capture wide-ranging momentary data from participants during their daily lives.77 DP measures fall into two broad categories: active or passive. Active DP requires participants to make deliberate responses on a smartphone or other portable device. For example, ecological momentary assessment (EMA), which has been extensively used in clinical psychology and psychiatry research,78–80 repeatedly cues participants to complete brief surveys about what they are doing, thinking, and feeling multiple times per day as they go about their regular activities. Participants can also be cued to complete performance-based measures, such as cognitive or social cognitive tasks.81,82
Passive DP measures require no deliberate action or effort on the part of the subject. Data are automatically extracted from sensors in smartphones or wearable devices as participants go about their daily activities. In addition, metadata on how participants interact with their smartphone or device, such as the number of texts/emails initiated and received, frequency of typing errors, linguistic analyses of message content, and time spent on social media, can be automatically captured. Other examples include actigraphy measures of movement, Global Positioning System (GPS) coordinates of location, physiological sensors to detect heart rate or electrodermal activity, and insole recordings of gait characteristics. The huge volume of data captured through these devices are transformed, using open-source or proprietary AI-based algorithms, into metrics with the potential for clinical meaningfulness (e.g., GPS coordinates can be quantified into the number of locations visited, distance traveled, or steps taken outside of one’s home).83 A proliferation of passive indices has emerged in the marketplace, and initial evidence increasingly supports their validity as indicators of specific signs/symptoms and functioning for many CNS conditions, including mood, anxiety, psychotic disorders, and dementia.77,84–87
DP technologies represent a new class of endpoints that have the potential to mitigate placebo response in several ways, although implementation in CNS clinical trials is just beginning. Several features of DP could reduce Category 2-related error associated with conventional ClinROs or PROs. For example, EMA can provide momentary, high-frequency assessments of daily activities and experiences while minimizing errors in retrospective reporting or recall biases. In line with this notion, a recent treatment study of distressed older adults found that EMA measures of depression and anxiety were more sensitive to treatment effects than conventional cross-sectional measures with longer recall periods.88,89 DP could also reduce Category 1 placebo response by reducing the subject-staff interactions required for outcome measurement. Since active DP measures are completed outside of a trial site, interactions with the site staff and trial environment are minimized. Patients might also provide more accurate reports for EMA than face-to-face ratings for several reasons, such as the relative anonymity of completing digital surveys; the fact that momentary response requirements preclude participants from having the opportunity to consider what they would be “expected” to report; and the potential for contemporaneous reports of behaviors and moods/symptoms to be able to adjust for common biases to report greater impairment and disability during periods of anxious or sad moods, which is a particular problem in trials with only a few dispersed assessment visits. Supporting these ideas, there is some evidence that people are more likely to report suicidal behavior with remote than in-person assessments.90 Furthermore, since passive DP assessments involve no subjective reporting, effort, or even awareness of the data collection on the subject’s part, one would expect them to be less susceptible to interpersonal and psychological factors that inflate placebo response.
DP can be integrated into clinical trials to mitigate placebo response in many ways. Two examples include assessing for nonadherence prior to randomization and conducting interim assessments between key in-person study visits.
Using DP to assess adherence before randomization. It is important to determine if potential clinical trial participants are unlikely to adhere to study procedures and will be at high risk to drop out. Identification strategies need to balance timing (as early as possible; well prior to randomization) and bias (do not exclude patients because of their critical target symptoms). During run-in periods prior to any blinded treatment, participants can be asked to participate in minimally demanding assessments that are not directly related to the primary outcomes of the study (e.g., answering a 2-question, 30-second, EMA survey querying location and social context) to test for longer-term adherence to study procedures. Available data in neuropsychiatric samples (e.g., schizophrenia, bipolar disorder) indicate that potentially nonadherent participants can be identified early and clinical symptom severity does not predict those patients found early to be at high risk for nonadherence,91 meaning that meeting severity criteria for a trial does not bias determinations of potential nonadherence.
Technology allows for dense repetition of assessments prior to initiation of treatment, which can probe for participants who manifest improvements or excessive variance in their reported symptoms across successive assessments. As these assessments can be initiated during screening periods prior to randomization, exclusion of participants who are excessively variable, responsive, or nonadherent does not involve retroactive elimination of randomized participants. Several large-scale studies have demonstrated very high correlations between early adherence to EMA assessments and eventual adherence over the course of the entire study.91
There are several factors associated with adherence to EMA assessments. Interestingly, compensation for participation is very important, with the least adherence in studies with no compensation, better adherence in studies with compensation, and the best adherence in studies with momentary (survey by survey) compensation.92 While compensation for engaging in device-based training, such as computerized cognitive training, might challenge the results of studies, participants who are assessed with many common EMA assessment strategies do not know what the “correct” answer is. The density of surveys is also important, with the paradoxical finding that more surveys per day is associated with greater adherence, up to a certain point.91,92 A once-per-day survey schedule means that a missed survey is a missed assessment day, while more dense survey strategies lessen the impact of individual missed surveys.
If one of the benefits of DP during a run-in period is to identify erratic, partially adherent, and potentially placebo-responding patients, what should be done when they are detected? Early and complete nonadherence to assessment procedures should lead to exclusion from the trial, particularly if this behavior is detected prior to randomization and the data are not shared concurrently with the site. Similarly, a placebo response induced by repeated assessments prior to any treatment also suggests that exclusion should be considered.
Using DP for interim assessments. Since exposure to excessive numbers of human interactions might produce Category 1 placebo responses, technology can also be used to perform interim assessments. It is likely true that assessments that are too widely separated might be associated with reduced engagement on the part of participants; interim assessments, even if the content is not obviously related to the primary outcome measure, could be a factor in increasing engagement in the study and increasing motivation on the part of participants to validly report their symptoms and functioning during in-person assessments. For example, in the SERENITY III Part 2 study by Bioexcel,93 participants with schizophrenia or bipolar disorder will be treated for agitation at home in a double-blind, placebo-controlled study. Participants and informants will report on the occurrence and severity of agitated episodes using EMA surveys. As agitated episodes are expected to be dispersed, informants and participants will be contacted daily and asked to answer agitation and mood questions to promote engagement in the study between agitated episodes.
Potential pitfalls of DP. Despite the appeal of DP, experience with these methods is limited, and their psychometric properties and validity for use in CNS trials is not adequately understood. There are several areas of potential concern, but results are inconsistent across studies, with some seeming much better than others. For example, correlations between legacy COAs and DP measures, or between active and passive DP measures, are sometimes vanishingly small (or counterintuitively related), raising questions about which metric is correct. There are several studies where correlations were found to be quite substantial,92 raising concerns about the sources of variation in convergence across studies.
It is also important to consider potential unintended, or even placebo-genic, consequences of DP assessments from the outset. For example, although DP decreases direct contact with the clinical site environment, people can have high levels of affinity for their digital devices, hold strong expectations about their capabilities, and even experience smartphone separation anxiety, suggesting that our devices might be infused with psychological meaning rather than being neutral objects.94 It is unclear if a device provided to a research participant would induce the same connection. Notably, although technology has the potential advantage of avoidance of the therapeutic benefits of empathy associated with human contact, it still involves exposure to assessment content. For example, several studies of posttraumatic stress disorder (PTSD) have had substantial placebo effects,95 which could be due to the exposure-related features of common PTSD outcome assessments; re-exposure during assessment of traumatic experiences and emotional reactions to those experiences could easily occur with technology as well.
Theoretically, completing multiple EMA surveys per day could induce a therapeutic effect, leading to reductions in symptoms even in the absence of a formal intervention.96–99 However, several studies have found essentially no changes in the severity of psychotic or mood disorder symptoms, with as many as 90 surveys over 30 days.92 The novelty of using a new device or app might strongly impact participants’ positive behavioral and experience changes apart from any active treatment-related effects.92
DP approaches might also have unintended adverse effects on trial participants. For example, repeated EMA queries about negative mood or distressing thoughts could lead to an unintended focus on and worsening of these symptoms. Notably, there is some evidence that asking people about suicide on EMA surveys does not appear to be a risk factor for increasing suicidal ideation,100 and that participants are willing to endorse suicidal ideation and related content in an EMA survey format.101,102 Furthermore, once the initial novelty of a new technology subsides, continued engagement of trial participants could be impacted. Past a certain point, lessening human interaction could have the unintended consequence of diminished participant engagement in trials and higher dropout rates.103 However, this is speculation, and in some studies, EMA measurements of symptoms and functioning have gone on for 12 months with minimal evidence of either participant study fatigue or deterioration in quality of data.
Conclusion and Paths Forward
A large range of available and rapidly emerging technologies have the potential to mitigate distinct components of the placebo response in CNS trials. However, the evidence base supporting their ability to meaningfully reduce placebo response is currently small. No published studies have directly evaluated whether any of the technologies reviewed demonstrate an impact on the placebo response in an actual clinical trial in which subjects are randomized to PRMT versus a control group. Of all the PRMTs considered, the most methodologically rigorous evaluations have been applied to participant training approaches and remote assessments. There is consistent support for novel training strategies based on a small set of studies; only one out of three relatively rigorous comparisons between remote- and site-based assessments demonstrated evidence of placebo response reduction. The promise of AI methods, genetic biomarkers, and blinded signal analytics comes from post hoc analyses. Furthermore, the potential positive or negative impacts of many newer technologies, such as direct outreach/advertising through social media and high frequency DP, have received minimal consideration. Overall, much more research, particularly prospective studies with randomized designs, is needed to convincingly demonstrate the efficacy of the technologies considered. There are several FDA-regulated studies in progress where this strategy is in place; as some of these studies are quite large, they might provide considerable evidence in the near future.
Products that aim to be marketed as PRMTs will require a higher standard of evidence to demonstrate placebo response reduction efficacy and should consider seeking formal approval as a medical device (e.g., if the product meets the definition of a medical device) or regulatory qualification via programs such as the FDA Qualification Program104 or the European Medicines Agency (EMA) qualification procedure.105 To make progress in addressing the pervasive placebo response challenge, the CNS field will benefit from pursuing additional pathways and collaborative efforts. One approach is for precompetitive collaboration among pharmaceutical companies to pool data and financial resources to develop and test promising solutions for the shared challenge of placebo response. For example, organizations like the Critical Path Institute (https://c-path.org) provide infrastructure to create a neutral environment for industry and academia, as well as regulators and other government agencies, to work together to accelerate and derisk drug development. Sharing and pooling data to compare results across completed trials using different technologies (e.g., versus conventional approaches) could also help guide the field toward promising approaches that warrant more rigorous evaluation.
Regulatory considerations. Like all aspects of drug development, PRMTs employed as part of new drug and device applications will encounter regulatory scrutiny. Looking ahead, it can be expected that more technologies aimed at mitigating placebo response, with varying levels of empirical support, will be incorporated into protocols. An increase in new technologies aimed at increasing trial efficiency that might inadvertently impact placebo response (positively or negatively) can also be expected. Since regulators have extensive experience with reviewing methods aimed at preventing error, we expect technologies directed at Category 2 placebo responses will raise less concern than those aimed at the mitigation of Category 1 placebo responses.
Regulators long accustomed to studies using single- and double-blind lead-in might recognize technologies that enhance blind and refine strategies as extensions of legacy experience. Proposals to predict placebo response to restrict the population studied will likely face a higher burden and/or more restrictions in the pursuit of regulatory acceptance, given potential questions regarding the generalizability of findings to the broader target patient population. This concern will likely be mitigated if the discovery of these risk factors, such as nonadherence, occurs very early in the screening process. Regulators do not require that a proportion of participants enrolled in clinical trials be nonadherent. It is important to consider that there are many situations wherein the selection process for clinical trials (e.g., exclusion of comorbidity in domains of substance use or obesity) makes the trial population less representative than the general population.
As investigators using PRMTs prepare to engage with regulators, it will be critical to understand the proposed technology, its operational details, and its intended context of use. Communication with regulators is key and most helpful when begun early. This could be especially important when the PRMT represents an addition to a protocol in a program that has previously failed due to high placebo response.
We conclude by highlighting three key questions. First, how should AI/ML algorithms be applied to identify placebo responders in clinical trials? The number of regulatory submissions to the FDA that included AI/ML approaches for drug development has grown considerably in the past five years.106 AI/ML is central to many technologies aimed at prospectively identifying placebo responders based on individual-level characteristics and blinded signal analytics, as well as DP. In 2021, the FDA and other regulatory agencies jointly issued 10 guiding principles for Good Machine Learning Practice for medical device measurement.107 Many of these principles, such as multidisciplinary collaboration, data quality assurance and robust security, and independence of training and testing datasets, are also applicable to drug development. For AI/ML applications in drug development, additional issues include standards for proper validation, locked versus continuously learning algorithms, generalizability, transparency/explainability (particularly for proprietary algorithms), and safety and privacy risks.108 The FDA subsequently published a discussion paper and request for feedback on a proposed regulatory framework for AI/ML-based software as a medical device and draft guidance on marketing submission recommendations for a predetermined change control plan for AI/ML-enabled device software functions.109,110 The EMA also recently published a draft reflection manuscript on this topic.111
Second, if we could prospectively identify placebo responders, how should that information be used to design and analyze data from clinical trials? One possibility would be a predictive sample enrichment approach that selects or stratifies patients based on placebo responder status, using prospectively planned and fixed criteria, with the expectation that the detection of a drug effect would be more likely than it would in an unselected population. From a scientific perspective, it is worth noting that the same processes that underlie placebo response might be an important component of the beneficial response experienced by patients receiving active treatment with the investigational product, raising the possibility that such approaches could diminish the separation of active versus placebo treatments.3,112 Aside from this consideration, general FDA guidance on enrichment strategies113 emphasizes concerns related to the generalizability and applicability of study results. To enrich for placebo nonresponders, sponsors would need to consider whether an enrichment approach based on placebo response status could be used in practice to identify patients to whom a drug should be given, which has key implications for drug labeling, as well as the extent to which patients who do not meet the selection criteria (i.e., placebo responders) should be studied. Furthermore, the accuracy of measurements to identify an enriched population is also key, yet we currently know little about the prevalence and sensitivity/specificity of AI/ML- or biomarker-based approaches to identifying placebo responders.
Third, how can DP be optimally leveraged to help mitigate the placebo response? The hope is that DP will address long-standing challenges, such as the placebo response, by virtue of superior validity and reliability, as well as decreased susceptibility to psychosocial influences, compared to legacy clinical phenotypes. Our understanding of the impact of DP on the placebo response is only just beginning to be informed by data. Recognizing the promise and rapid expansion of DP technologies, the FDA recently issued draft guidance on the use of digital health technologies to acquire data remotely.114 The document describes key regulatory considerations for selecting measures that are fit for purpose, with demonstrated psychometrics, validity, and usability in the target population. Digital technologies also raise unique issues related to data integrity, interoperability, cybersecurity/hacking risks, and attributability; many of these issues are addressed in the recently published EMA Guideline on Computerized Systems and Electronic Data in Clinical Trials.115 The FDA has recently endorsed Phase III research designs in which DP is a central element of either subject selection or data collection.51,116 Thus, some DP technologies are now transparent, reliable, and valid enough for regulators to understand their procedures and outcomes and agree with their purpose in the trials. Stakeholders need to continue developing policies and procedures to address these issues.117,118
The placebo response challenge in CNS trials has a long and well-documented history. Although a range of current and emerging technologies show promise for addressing this challenge, the evidence base is small to date. Meaningful progress will require careful consideration of the potential pitfalls of rapidly emerging technologies, rigorous evaluation of their putative effects, and close collaboration among diverse stakeholders committed to CNS drug discovery.
Disclaimer
This manuscript is the work product of the the International Society for CNS Clinical Trials and Methodology (ISCTM) Innovative Technologies for CNS Clinical Trials working group chaired by MD and RSEK that began at the 15th Annual ISCTM Scientific Meeting in February 2019 in Washington, DC. This article reflects the views of the authors and should not be understood or quoted as being made on behalf or reflecting the position of the agencies or organizations with which the author(s) is/are employed/affiliated.
References
- Kinon BJ, Potts AJ, Watson SB. Placebo response in clinical trials with schizophrenia patients. Curr Opin Psychiatry. 2011;24(2):107–113.
- Rutherford BR, Pott E, Tandler JM, et al. Placebo response in antipsychotic clinical trials: a meta-analysis. JAMA Psychiatry. 2014;71(12):1409–1421.
- Rief W, Nestoriuc Y, Weiss S, et al. Meta-analysis of the placebo response in antidepressant trials. J Affect Disord. 2009;118(1–3):1–8.
- Khan A, Brown WA. Antidepressants versus placebo in major depression: an overview. World Psychiatry. 2015;14(3):294–300.
- Gopalakrishnan M, Zhu H, Farchione TR, et al. The trend of increasing placebo response and decreasing treatment effect in schizophrenia trials continues: an update from the US Food and Drug Administration. J Clin Psychiatry. 2020;81(2):19r12960.
- Khan A, Fahl Mar K, Faucett J, et al. Has the rising placebo response impacted antidepressant clinical trial outcome? Data from the US Food and Drug Administration 1987–2013. World Psychiatry. 2017;16(2):181–192.
- Leucht S, Heres S, Davis JM. Increasing placebo response in antipsychotic drug trials: let’s stop the vicious circle. Am J Psychiatry. 2013;170(11):1232–1234.
- Salloum NC, Fava M, Ball S, Papakostas GI. Success and efficiency of Phase 2/3 adjunctive trials for MDD funded by industry: a systematic review. Mol Psychiatry. 2020;25(9):1967–1974.
- van Gerven J, Cohen A. Vanishing clinical psychopharmacology. Br J Clin Pharmacol. 2011;72(1):1–5.
- Fava M. The role of regulators, investigators, and patient participants in the rise of the placebo response in major depressive disorder. World Psychiatry. 2015;14(3):307–308.
- Fava GA, Ruini C, Sonino N. Treatment of recurrent depression: a sequential psychotherapeutic and psychopharmacological approach. CNS Drugs. 2003;17(15):1109–1117.
- Papakostas GI, Ostergaard SD, Iovieno N. The nature of placebo response in clinical studies of major depressive disorder. J Clin Psychiatry. 2015;76(4):456–466.
- Inan OT, Tenaerts P, Prindiville SA, et al. Digitizing clinical trials. NPJ Digit Med. 2020;3:101.
- Khozin S, Coravos A. Decentralized trials in the age of real-world evidence and inclusivity in clinical investigations. Clin Pharmacol Ther. 2019;106(1):25–27.
- Wager TD, Atlas LY. The neuroscience of placebo effects: connecting context, learning and health. Nat Rev Neurosci. 2015;16(7):403–418.
- Enck P, Klosterhalfen S. Placebos and the placebo effect in drug trials. Handb Exp Pharmacol. 2019;260:399–431.
- Benedetti F, Carlino E, Piedimonte A. Increasing uncertainty in CNS clinical trials: the role of placebo, nocebo, and Hawthorne effects. Lancet Neurol. 2016;15(7):736–747.
- Kaptchuk TJ, Kelley JM, Conboy LA, et al. Components of placebo effect: randomised controlled trial in patients with irritable bowel syndrome. BMJ. 2008;336(7651):999–1003.
- Czerniak E, Biegon A, Ziv A, et al. Manipulating the placebo response in experimental pain by altering doctor’s performance style. Front Psychol. 2016;7:874.
- von Wernsdorff M, Loef M, Tuschen-Caffier B, Schmidt S. Effects of open-label placebos in clinical trials: a systematic review and meta-analysis. Sci Rep. 2021;11(1):3855.
- Kaptchuk TJ, Kelley JM, Deykin A, et al. Do “placebo responders” exist? Contemp Clin Trials. 2008;29(4):587–595.
- Horing B, Weimer K, Muth ER, Enck P. Prediction of placebo responses: a systematic review of the literature. Front Psychol. 2014;5:1079.
- Zilcha-Mano S, Roose SP, Brown PJ, Rutherford BR. A machine learning approach to identifying placebo responders in late-life depression trials. Am J Geriatr Psychiatry. 2018;26(6):669–677.
- Trivedi MH, South C, Jha MK, et al. A novel strategy to identify placebo responders: prediction index of clinical and biological markers in the EMBARC trial. Psychother Psychosom. 2018;87(5):285–295.
- Lorenzo-Luaces L, Rodriguez-Quintana N, Riley TN, Weisz JR. A placebo prognostic index (PI) as a moderator of outcomes in the treatment of adolescent depression: could it inform risk-stratification in treatment with cognitive-behavioral therapy, fluoxetine, or their combination? Psychother Res. 2021;31(1):5–18.
- Zilcha-Mano S, Brown PJ, Roose SP, et al. Optimizing patient expectancy in the pharmacologic treatment of major depressive disorder. Psychol Med. 2019;49(14):2414–2420.
- Zilcha-Mano S, Roose SP, Brown PJ, Rutherford BR. Abrupt symptom improvements in antidepressant clinical trials: transient placebo effects or therapeutic reality? J Clin Psychiatry. 2018;80(1):18m12353.
- Sikora M, Heffernan J, Avery ET, et al. Salience network functional connectivity predicts placebo effects in major depression. Biol Psychiatry Cogn Neurosci Neuroimaging. 2016;1(1):68-76.
- Tu Y, Ortiz A, Gollub RL, et al. Multivariate resting-state functional connectivity predicts responses to real and sham acupuncture treatment in chronic low back pain. Neuroimage Clin. 2019;23:101885. Erratum in: Neuroimage Clin. 2019;24:102105.
- Smith EA, Horan WP, Demolle D, et al. Using artificial intelligence-based methods to address the placebo response in clinical trials. Innov Clin Neurosci. 2022;19(1–3):60–70.
- Hall KT, Kaptchuk TJ. Genetic biomarkers of placebo response: what could it mean for future trial design? Clin Investig (Lond). 2013;3(4):311–314.
- Hall KT, Loscalzo J, Kaptchuk TJ. Systems pharmacogenomics – gene, disease, drug and placebo interactions: a case study in COMT. Pharmacogenomics. 2019;20(7):529–551.
- Hall KT, Loscalzo J, Kaptchuk TJ. Genetics and the placebo effect: the placebome. Trends Mol Med. 2015;21(5):285–294.
- Zubieta JK, Stohler CS. Neurobiological mechanisms of placebo responses. Ann N Y Acad Sci. 2009;1156:198–210.
- Benedetti F, Amanzio M. Mechanisms of the placebo response. Pulm Pharmacol Ther. 2013;26(5):520-523.
- Scott DJ, Stohler CS, Egnatuk CM, et al. Placebo and nocebo effects are defined by opposite opioid and dopaminergic responses. Arch Gen Psychiatry. 2008;65(2):220–231.
- Lidstone SC, Schulzer M, Dinelle K, et al. Effects of expectation on placebo-induced dopamine release in Parkinson disease. Arch Gen Psychiatry. 2010;67(8):857–865.
- Price DD, Milling LS, Kirsch I, et al. An analysis of factors that contribute to the magnitude of placebo analgesia in an experimental paradigm. Pain. 1999;83(2):147–156.
- Erpelding N, Evans K, Lanier RK, et al. Placebo response reduction and accurate pain reporting training reduces placebo responses in a clinical trial on chronic low back pain: results from a comparison to the literature. Clin J Pain. 2020;36(12):950–954.
- Katz N, Dworkin RH, North R, et al. Research design considerations for randomized controlled trials of spinal cord stimulation for pain: Initiative on Methods, Measurement, and Pain Assessment in Clinical Trials/Institute of Neuromodulation/International Neuromodulation Society recommendations. Pain. 2021;162(7):1935-1956.
- Kuperman P, Talmi D, Katz N, Treister R. Certainty in ascending sensory signals – the unexplored driver of analgesic placebo response. Med Hypotheses. 2020;143:110113.
- Treister R, Honigman L, Lawal OD, et al. A deeper look at pain variability and its relationship with the placebo response: results from a randomized, double-blind, placebo-controlled clinical trial of naproxen in osteoarthritis of the knee. Pain. 2019;160(7):1522-1528.
- Treister R, Lawal OD, Shecter JD, et al. Accurate pain reporting training diminishes the placebo response: results from a randomised, double-blind, crossover trial. PLoS One. 2018;13(5):e0197844.
- Backonja M, Williams L, Miao X, et al. Safety and efficacy of neublastin in painful lumbosacral radiculopathy: a randomized, double-blinded, placebo-controlled phase 2 trial using Bayesian adaptive design (the SPRINT trial). Pain. 2017;158(9):1802–1812.
- Cohen EA, Hassman HH, Ereshefsky L, et al. Placebo response mitigation with a participant-focused psychoeducational procedure: a randomized, single-blind, all placebo study in major depressive and psychotic disorders. Neuropsychopharmacology. 2021;46(4):844–850.
- United States Food and Drug Administration. Conduct of clinical trials of medical products during the COVID-19 public health emergency: guidance for industry, investigators, and institutional review boards. Aug 2021. https://collections.nlm.nih.gov/catalog/nlm:nlmuid-9918248910206676-pdf. Accessed 19 Feb 2024.
- Kobak KA, Leuchter A, DeBrota D, et al. Site versus centralized raters in a clinical depression trial: impact on patient selection and placebo response. J Clin Psychopharmacol. 2010;30(2):193–197.
- Targum SD, Wedel PC, Robinson J, et al. A comparative analysis between site-based and centralized ratings and patient self-ratings in a clinical trial of major depressive disorder. J Psychiatr Res. 2013;47(7):944–954.
- Williams JB, Kobak KA, Giller E, et al. Comparison of site-based versus central ratings in a study of generalized anxiety disorder. J Clin Psychopharmacol. 2015;35(6):654–660.
- Blier P, Szegedi A, Hayes R, et al. Rapastinel for the treatment of major depressive disorder: a patient-centric clinical development program. Poster presented at: The Canadian Psychiatric Associations’s (CPA) Annual Conference, 12–14 September 2019. Quebec City, Canada.
- Hauser RA, Factor SA, Marder SR, et al. KINECT 3: a Phase 3 randomized, double-blind, placebo-controlled trial of valbenazine for tardive dyskinesia. Am J Psychiatry. 2017;174(5):476–484.
- Faasse K, Grey A, Jordan R, et al. Seeing is believing: impact of social modeling on placebo and nocebo responding. Health Psychol. 2015;34(8):880-885.
- Kamenica E, Naclerio R, Malani A. Advertisements impact the physiological efficacy of a branded drug. Proc Natl Acad Sci U S A. 2013;110(32):12931–12935.
- Greenslit NP, Kaptchuk TJ. Antidepressants and advertising: psychopharmaceuticals in crisis. Yale J Biol Med. 2012;85(1):153–158.
- Darmawan I, Bakker C, Brockman TA, et al. The role of social media in enhancing clinical trial recruitment: scoping review. J Med Internet Res. 2020;22(10):e22810.
- Abujarad F, Peduzzi P, Mun S, et al. Comparing a multimedia digital informed consent tool with traditional paper-based methods: randomized controlled trial. JMIR Form Res. 2021;5(10):e20458.
- Chen C, Lee PI, Pain KJ, et al. Replacing paper informed consent with electronic informed consent for research in academic medical centers: a scoping review. AMIA Jt Summits Transl Sci Proc. 2020;2020:80–88.
- Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960;23(1):56–62.
- Sachs G, Peters A. What blinded raters don’t see: relapse events in a bipolar study detected by site-based raters versus a computer simulated rater. Abstract presented at: International Society for CNS Clinical Trials and Methodology 12th Annual Scientific Meeting. 16–18 Feb 2016. Washington, DC.
- Sachs G, DeBonis D, Wang X, Epstein B. Is a computer simulated rater good enough to administer the Hamilton Depression Rating Scale in clinical trials? Poster presented at: International Society for CNS Clinical Trials and Methodology Autumn Conference. 27–29 August 2015. Amsterdam, The Netherlands.
- Sachs GS, Vanderburg DG, Edman S, et al. Adjunctive oral ziprasidone in patients with acute mania treated with lithium or divalproex, part 2: influence of protocol-specific eligibility criteria on signal detection. J Clin Psychiatry. 2012;73(11):1420–1425.
- Opler MGA, Yavorsky C, Daniel DG. Positive and Negative Syndrome Scale (PANSS) training: challenges, solutions, and future directions. Innov Clin Neurosci. 2017;14(11–12):77–81.
- Gary ST, Otero AV, Falkner KG, Diaas NR. Validation and equivalency of electronic clinical outcomes assessment systems. Int J Clin Trials. 2020;7(4):271–277.
- Bain EE, Shafner L, Walling DP, et al. Use of a novel artificial intelligence platform on mobile devices to assess dosing compliance in a Phase 2 clinical trial in subjects with schizophrenia. JMIR Mhealth Uhealth. 2017;5(2):e18.
- Labovitz DL, Shafner L, Reyes Gil M, et al. Using artificial intelligence to reduce the risk of nonadherence in patients on anticoagulation therapy. Stroke. 2017;48(5):1416–1419.
- Reilly-Harrington NA, DeBonis D, Leon AC, et al. The interactive computer interview for mania. Bipolar Disord. 2010;12(5):521-527.
- Vaidyam AN, Linggonegoro D, Torous J. Changes to the psychiatric chatbot landscape: a systematic review of conversational agents in serious mental illness: Changements du paysage psychiatrique des chatbots: une revue systematique des agents conversationnels dans la maladie mentale serieuse. Can J Psychiatry. 2021;66(4):339–348.
- Torous J, Bucci S, Bell IH, et al. The growing field of digital psychiatry: current evidence and the future of apps, social media, chatbots, and virtual reality. World Psychiatry. 2021;20(3):318–335.
- Valmaggia LR, Latif L, Kempton MJ, Rus-Calafell M. Virtual reality in the psychological treatment for mental health problems: an systematic review of recent evidence. Psychiatry Res. 2016;236:189–195.
- Parsons TD. Virtual reality for enhanced ecological validity and experimental control in the clinical, affective and social neurosciences. Front Hum Neurosci. 2015;9:660.
- Freeman D, Reeve S, Robinson A, et al. Virtual reality in the assessment, understanding, and treatment of mental health disorders. Psychol Med. 2017;47(14):2393–2400.
- Bell IH, Nicholas J, Alvarez-Jimenez M, et al. Virtual reality as a clinical tool in mental health research and practice. Dialogues Clin Neurosci. 2020;22(2):169–177.
- Umbricht D, Kott A, Daniel DG. The effects of erratic ratings on placebo response and signal detection in the Roche Bitopertin Phase 3 negative symptoms studies—a post hoc analysis. Schizophr Bull Open. 2020;1(1):sgaa040.
- Targum SD, Pendergrass JC, Lee S, Loebel A. Ratings surveillance and reliability in a study of major depressive disorder with subthreshold hypomania (mixed features). Int J Methods Psychiatr Res. 2018;2(4)7:e1729.
- Targum SD, Cameron BR, Ferreira L, MacDonald ID. Early score fluctuation and placebo response in a study of major depressive disorder. J Psychiatr Res. 2020;121:118–125.
- Targum SD, Catania CJ. Audio-digital recordings for surveillance in clinical trials of major depressive disorder. Contemp Clin Trials Commun. 2019;14:100317.
- Harvey PD, Depp CA, Rizzo AA, et al. Technology and mental health: state of the art for assessment and treatment. Am J Psychiatry. 2022;179(12):897–914.
- Myin-Germeys I, Oorschot M, Collip D, et al. Experience sampling research in psychopathology: opening the black box of daily life. Psychol Med. 2009;39(9):1533–1547.
- Oorschot M, Kwapil T, Delespaul P, Myin-Germeys I. Momentary assessment research in psychosis. Psychol Assess. 2009;21(4):498–505.
- Shiffman S, Stone AA, Hufford MR. Ecological momentary assessment. Annu Rev Clin Psychol. 2008;4:1–32.
- Weizenbaum E, Torous J, Fulford D. Cognition in context: understanding the everyday predictors of cognitive performance in a new era of measurement. JMIR Mhealth Uhealth. 2020;8(7):e14328.
- Weizenbaum EL, Fulford D, Torous J, et al. Smartphone-based neuropsychological assessment in Parkinson’s disease: feasibility, validity, and contextually driven variability in cognition. J Int Neuropsychol Soc. 2022;28(4):401–413.
- Koppe G, Meyer-Lindenberg A, Durstewitz D. Deep learning for small and big data in psychiatry. Neuropsychopharmacology. 2021;46(1):176–190.
- Dogan E, Sander C, Wagner X, et al. Smartphone-based monitoring of objective and subjective data in affective disorders: where are we and where are we going? Systematic review. J Med Internet Res. 2017;19(7):e262.
- Barnett I, Torous J, Staples P, et al. Relapse prediction in schizophrenia through digital phenotyping: a pilot study. Neuropsychopharmacology. 2018;43(8):1660–1666.
- Benoit J, Onyeaka H, Keshavan M, Torous J. Systematic review of digital phenotyping and machine learning in psychosis spectrum illnesses. Harv Rev Psychiatry. 2020;28(5):296–304.
- Piau A, Wild K, Mattek N, Kaye J. Current state of digital biomarker technologies for real-life, home-based monitoring of cognitive function for mild cognitive impairment to mild Alzheimer disease and implications for clinical care: systematic review. J Med Internet Res. 2019;21(8):e12785.
- Moore RC, Depp CA, Wetherell JL, Lenze EJ. Ecological momentary assessment versus standard assessment instruments for measuring mindfulness, depressed mood, and anxiety among older adults. J Psychiatr Res. 2016;75:116–123.
- Targum SD, Sauder C, Evans M, et al. Ecological momentary assessment as a measurement tool in depression trials. J Psychiatr Res. 2021;136:256–264.
- Torous J, Staples P, Shanahan M, et al. Utilizing a personal smartphone custom app to assess the Patient Health Questionnaire-9 (PHQ-9) depressive symptoms in patients with major depressive disorder. JMIR Ment Health. 2015;2(1):e8.
- Jones SE, Moore RC, Pinkham AE, et al. A cross-diagnostic study of adherence to ecological momentary assessment: comparisons across study length and daily survey frequency find that early adherence is a potent predictor of study-long adherence. Pers Med Psychiatry. 2021;29–30:100085.
- Harvey PD, Miller ML, Moore RC, et al. Capturing clinical symptoms with ecological momentary assessment: convergence of momentary reports of psychotic and mood symptoms with diagnoses and standard clinical assessments. Innov Clin Neurosci. 2021;18(1–3):24–30.
- Clinicaltrials.gov. Dexmedetomidine in the treatment of agitation associated with schizophrenia and bipolar disorder (SERENITY III). https://clinicaltrials.gov/study/NCT05658510. Accessed 14 Dec 2023.
- Clayton RB, Leshner G, Almond A. The extended iSelf: the impact of iPhone separation on cognition, emotion, and physiology. J Comput-Mediat Comm. 2015;20(2):119–135.
- Hodgins GE, Blommel JG, Dunlop BW, et al. Placebo effects across self-report, clinician rating, and objective performance tasks among women with post-traumatic stress disorder: investigation of placebo response in a pharmacological treatment study of post-traumatic stress disorder. J Clin Psychopharmacol. 2018;38(3):200–206.
- Kauer SD, Reid SC, Crooke AH, et al. Self-monitoring using mobile phones in the early stages of adolescent depression: randomized controlled trial. J Med Internet Res. 2012;14(3):e67.
- Dubad M, Elahi F, Marwaha S. The clinical impacts of mobile mood-monitoring in young people with mental health problems: the MeMO Study. Front Psychiatry. 2021;12:687270.
- Firth J, Torous J, Nicholas J, et al. The efficacy of smartphone-based mental health interventions for depressive symptoms: a meta-analysis of randomized controlled trials. World Psychiatry. 2017;16(3):287–298.
- Liu JY, Xu KK, Zhu GL, et al. Effects of smartphone-based interventions and monitoring on bipolar disorder: a systematic review and meta-analysis. World J Psychiatry. 2020;10(11):272–285.
- Husky M, Olie E, Guillaume S, et al. Feasibility and validity of ecological momentary assessment in the investigation of suicide risk. Psychiatry Res. 2014;220(1–2):564–570.
- Lieberman A, Parrish EM, Depp CA, et al. Demoralization in schizophrenia: a pathway to suicidal ideation? Arch Suicide Res. 2023:1–15. Epub ahead of print.
- Parrish EM, Chalker SA, Cano M, et al. Ecological momentary assessment of interpersonal theory of suicide constructs in people experiencing psychotic symptoms. J Psychiatr Res. 2021;140:496–503.
- Henson P, Wisniewski H, Hollis C, et al. Digital mental health apps and the therapeutic alliance: initial review. BJPsych Open. 2019;5(1):e15.
- United States Food and Drug Administration. Clinical outcome assessments (COA) qualification program. Current as of 27 Oct 2023. https://www.fda.gov/drugs/drug-development-tool-ddt-qualification-programs/clinical-outcome-assessment-coa-qualification-program. Accessed 14 Dec 2023.
- European Medicines Agency. Qualification of novel methodologies for drug development: guidance to applicant. 10 Nov 2014. Revised Oct 2023. https://www.ema.europa.eu/en/documents/regulatory-procedural-guideline/qualification-novel-methodologies-drug-development-guidance-applicants_en.pdf. Accessed 14 Dec 2023.
- Liu Q, Huang R, Hsieh J, et al. Landscape analysis of the application of artificial intelligence and machine learning in regulatory submissions for drug development from 2016 to 2021. Clin Pharmacol Ther. 2023;113(4):771–774.
- United Staters Food and Drug Administration. Good machine learning practice for medical device development: guiding principles. Current as of 27 Oct 2021. https://www.fda.gov/medical-devices/software-medical-device-samd/good-machine-learning-practice-medical-device-development-guiding-principles. Accessed 14 Dec 2023.
- Liu Q, Zhu H, Liu C, et al. Application of machine learning in drug development and regulation: current status and future potential. Clin Pharmacol Ther. 2020;107(4):726–729.
- United States Food and Drug Administration. Proposed regulatory framework for modifications to artificial intelligence/machine learning (AI/ML)-based software as a medical device (SaMD). Current as of 22 Sep 2021. https://www.fda.gov/medical-devices/software-medical-device-samd/artificial-intelligence-and-machine-learning-software-medical-device. Accessed 14 Dec 2023.
- United States Food and Drug Administration. Marketing submission recommendations for a predetermined change control plan for artificial intelligence/machine learning (AI/ML)-enabled device software functions: draft guidance for industry and Food and Drug Administration staff. Apr 2023. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/marketing-submission-recommendations-predetermined-change-control-plan-artificial. Accessed 14 Dec 2023.
- European Medicines Agency. Reflection paper on the use of artificial intelligence in the lifecycle of medicines. 19 Jul 2023. https://www.ema.europa.eu/en/news/reflection-paper-use-artificial-intelligence-lifecycle-medicines. Accessed 14 Dec 2023.
- Hauser W, Bartram-Wunn E, Bartram C, et al. Systematic review: placebo response in drug trials of fibromyalgia syndrome and painful peripheral diabetic neuropathy-magnitude and patient-related predictors. Pain. 2011;152(8):1709–1717.
- United States Food and Drug Administration. Enrichment strategies for clinical trials to support approval of human drugs and biological products. Mar 2019. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/enrichment-strategies-clinical-trials-support-approval-human-drugs-and-biological-products. Accessed 14 Dec 2023.
- United States Food and Drug Administration. Digital health technologies for remote data acquisition in clinical investigations: draft guidance for industry, investigators, and other stakeholders. Dec 2021. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/digital-health-technologies-remote-data-acquisition-clinical-investigations. Accessed 14 Dec 2023.
- European Medicines Agency. Guideline on computerised systems and electronic data in clinical trials. 9 Mar 2023. https://www.ema.europa.eu/en/documents/regulatory-procedural-guideline/guideline-computerised-systems-electronic-data-clinical-trials_en.pdf. Accessed 14 Dec 2023.
- GlobeNewswire. ANeuroTech receives IND approval from the FDA for pivotal Phase IIIB trial of adjunctive anti-depression drug, ANT-01. 5 Jun 2023. https://www.globenewswire.com/news-release/2023/06/05/2681798/0/en/ANeuroTech-receives-IND-approval-from-the-FDA-for-pivotal-Phase-IIIB-trial-of-adjunctive-anti-depression-drug-ANT-01.html. Accessed 14 Dec 2023.
- Byrom B, Watson C, Doll H, et al. Selection of and evidentiary considerations for wearable devices and their measurements for use in regulatory decision making: recommendations from the ePRO Consortium. Value Health. 2018;21(6):631–639.
- Walton MK, Cappelleri JC, Byrom B, et al. Considerations for development of an evidence dossier to support the use of mobile sensor technology for clinical outcome assessments in clinical trials. Contemp Clin Trials. 2020;91:105962.