

by Laetitia Pouzol, MSc, PharmD; Luca Piali, PhD; Claude CA Bernard, PhD; Marianne M. Martinic, PhD; Beat Steiner, PhD; and Martine Clozel, MD
Drs. Pouzol and Clozel were with Actelion Pharmaceuticals Ltd. in Allschwil, Switzerland, at the time of this research, and are presently with Idorsia Pharmaceuticals Ltd. in Allschwil, Switzerland. Dr. Piali was with Actelion Pharmaceuticals Ltd. in Allschwil, Switzerland, at the time of this research, and is presently with Hoffmann la Roche in Basel, Switzerland. Dr. Bernard is with Monash University, Faculty of Medicine, Nursing & Health Sciences in Melbourne, Australia. Dr. Martinic is with Idorsia Pharmaceuticals Ltd. in Allschwil, Switzerland. Dr. Steiner was with Actelion Pharmaceuticals Ltd. in Allschwil, Switzerland, at the time of this research.
Funding: No funding was provided.
Disclosures: The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Drs. Pouzol, Piali, Steiner, and Clozel acknowledge that the work was performed as employees of Actelion Pharmaceuticals Ltd. During manuscript preparation, Actelion Pharmaceuticals Ltd. was acquired by Johnson & Johnson, and its drug discovery and early development activities subsequently transferred into a newly created company, Idorsia Pharmaceuticals Ltd.
Innov Clin Neurosci. 2019;16(3–4):22–30
Abstract: Background: Despite the recent approval of new oral therapies for the treatment of multiple sclerosis (MS), a significant percentage of patients are still not free from disease activity. In view of the complex pathogenesis and the relapsing and progressive nature of MS, combination therapy, a classical approach to treat many chronic diseases, could improve disease control over monotherapy. Ponesimod, a selective and rapidly reversible sphingosine-1-phosphate receptor Type 1 (S1P1) modulator, currently in Phase III clinical trial stage in relapsing MS (RMS), and dimethyl fumarate (DMF) would potentially be an ideal combination due to their differing mechanisms of action and oral administration. Objective: The goal of the study was to evaluate the therapeutic effect of ponesimod monotherapy and investigate the potential additive, or synergistic, activity of ponesimod-DMF combination therapy in experimental autoimmune encephalomyelitis (EAE) animal models of MS. Methods: Efficacy was evaluated in the myelin oligodendrocyte glycoprotein (MOG)-induced EAE model in C57BL/6 mice (ponesimod monotherapy) and in the myelin basic protein (MBP)-induced EAE model in Lewis rats (monotherapies and combination therapy). The principal readout was the clinical score assessing paralysis. Additional readouts, such as histopathology, survival, and disease prevalence, were generated in parallel when applicable. Results: Ponesimod monotherapy in the mouse EAE model showed significant efficacy in both preventative and therapeutic settings. In the rat EAE model, ponesimod demonstrated significant dose-dependent efficacy on clinical scores, while DMF showed only modest activity. Combination therapy synergistically reduced the severity and prevalence of disease. Only the combination treatment of ponesimod and DMF fully suppressed clinical disease activity by the end of the study. Conclusion: The results support the potential therapeutic benefits of combining ponesimod with DMF to improve disease activity control in patients with MS. Additionally, the results suggest that combining ponesimod with other oral agents that have different mechanisms of action might also be therapeutically beneficial to patients with MS.
Keywords: Experimental autoimmune encephalomyelitis (EAE), multiple sclerosis (MS), dimethyl fumarate (DMF), sphingosine-1-phosphate, combination therapy, disease activity
The arrival of oral therapies for the treatment of multiple sclerosis (MS) has represented a significant medical breakthrough for the two million-plus people worldwide who have MS.1 Fingolimod was approved in 2010, followed by teriflunomide in 2012, and dimethyl fumarate (DMF) in 2013. These drugs have entered the MS treatment landscape, offering different efficacy profiles and improvements in therapeutic adherence.2 Despite these new treatments, however, curative therapies for MS remain elusive, and a significant proportion of patients still experience clinical relapses and disability progression.3–5 “No evidence of disease activity” (NEDA), the emerging endpoint in the treatment of MS, remains an unmet need, and new therapeutic strategies are needed. Although combination therapies are the standard of practice in other chronic progressive diseases, to date no routinely used therapeutic combination protocol for MS exists.
Based on promising efficacy and safety results from a Phase II study, ponesimod, a selective and rapidly reversible modulator of the sphingosine-1-phosphate receptor type 1 (S1P1), is currently in Phase III clinical trial stage for the treatment of relapsing MS (RMS).6 Ponesimod causes internalization of S1P1 receptors on lymphocytes, which results in blocking lymphocyte egress from lymphoid tissues into the circulation, thus reducing peripheral lymphocyte count in a dose-dependent manner in animals and humans.6–8 Oral ponesimod reduces inflammation and clinical disease parameters, leading to a maximal reduction of lymphocyte count over 24 hours in autoimmune disease models.8 Upon ponesimod treatment, pathogenic lymphocytes are sequestered in peripheral lymphoid organs, thus preventing their infiltration into the site of inflammation (i.e., the central nervous system [CNS] in MS).9 However, this mechanism of action does not affect other white blood cell types and should not impact antigen-dependent T cell activation, expansion, and survival.10 In addition to the effect on lymphocyte trafficking, preclinical data has suggested that S1P receptor modulators might have a direct beneficial effect on CNS resident cells, such as astrocytes, oligodendrocytes, and neurons.11,12 The relevance of targeting S1P receptors in MS has been well established with fingolimod, a nonselective S1P modulator active on four S1P receptors (S1P1, 3–5) and the first oral treatment approved for RMS.13 Although the effect on lymphocyte egress out of the lymph nodes into the circulation is exclusively mediated by S1P1,14 the four other S1P receptors (S1P2–5) mediate a wide range of responses to S1P (e.g., effects on vasoconstriction,15 fibrosis, and other functions on different cells).16 With the exception of bradycardia, which is believed to be mediated via transient S1P1 activation,17 other common adverse effects observed with fingolimod, such as dyspnea, might be due to off-target effects via other S1P receptors. Ponesimod presents distinctive features compared to fingolimod, including selectivity for S1P1, a shorter half-life, and consequently a faster lymphocyte count normalization upon treatment discontinuation (within less than a week) in comparison to fingolimod (1–2 months).18,19 Moreover, an optimized dose titration regimen for ponesimod treatment initiation was found to minimize the first-dose cardiac effects due to the modulation of S1P1 receptor on cardiomyocytes.6 Ponesimod’s S1P1 selectivity and rapid lymphocyte count reversibility might therefore improve its overall safety and tolerability profile and make it an ideal combination partner with DMF or teriflunomide for treatment of MS.
DMF (BG-12, Tecfidera) is an oral fumaric acid ester approved by the United States Food and Drug Administration (FDA) for the treatment of RMS in 2013. Phase III clinical studies on DMF demonstrated a significant reduction in relapse rate and radiological signs of disease activity compared to placebo.4 Fumarates have been investigated as possible anti-inflammatory substances since the 1950s.20 However, the underlying mechanism for the therapeutic effect of DMF in MS is not fully understood. Preclinical studies have demonstrated that DMF exhibits anti-inflammatory properties by inhibiting the translocation of the transcription-factor NF-kappaB,21–24 a key inducer of pro-inflammatory cytokines and inhibitor of T cell apoptosis. Other studies have reported the alteration of dendritic cell polarization, resulting in a shift in the polarization of T helper (Th) cells toward a Th2 rather than a Th1 or Th17 phenotype.25–28 Recently, research has shown that DMF inhibits aerobic glycolysis in activated myeloid and lymphoid cells, shifting the immune response from an immunostimulatory to an immunoregulatory environment.29 DMF has also demonstrated antioxidant properties,30 and the DMF metabolite, monomethyl fumarate (MMF),31 is able to cross the blood brain barrier (BBB), supporting a direct effect on CNS-resident cells.32 Reduction of clinical scores by DMF in mouse models of experimental autoimmune encephalomyelitis (EAE) was achieved in both preventive and therapeutic settings.32–34 DMF reduced infiltration of macrophages/microglia into the spinal cord, increased the anti-inflammatory cytokine IL-10 in the acute phase of the model,33 and induced neuroprotective effects, such as preservation of myelin and axons and reduced astrogliosis in the chronic phase of disease. In contrast to S1P1 modulators, DMF treatment did not affect T cell infiltration into the spinal cord and brain.32
Due to the inherent complexity of MS and individualized treatment options that address particular pathophysiological pathways, we hypothesized that combining drugs with different mechanisms of action would yield improved clinical scores in models of MS, such as EAE. The pathophysiology of EAE, which resembles MS in many ways, is characterized by an increased BBB permeability, an inflammatory mononuclear cell infiltration into the CNS (which results in the activation of resident glial cells), and the secretion of inflammatory cytokines.6,33,35,36 These inflammatory events lead to CNS demyelination and, consequently, paralysis. Although one single model cannot exactly mimic all of the aspects of the human disease, EAE has been shown to be a powerful model to determine efficacy and identify mechanisms of action of immunomodulatory therapies in MS, such as DMF and fingolimod.32–34,37,38
Here, we address whether the oral S1P1 receptor modulator ponesimod ameliorates disease in two different EAE models and whether the combination of ponesimod with the neuroprotectant DMF leads to an enhanced control of rat EAE disease.
Materials and Methods
Animals and treatment administration. Female C57BL/6 mice weighing 20 to 22g were obtained from Monash Animal Services, Australia, or Harlan Netherlands. Studies performed at LaTrobe University in Australia were in accordance with the Australian National Health and Medical Research Council’s Code of Practice for the Care and Use of Animals for Scientific Purposes (1997), after approval by Monash University Animal Ethics Committee (Clayton/Melbourne, Australia). Mice were included in the experiments after the acclimatization period (7–10 days).
Female Lewis rats weighing 180 to 200g at study initiation were purchased from Envigo Laboratory Rx, Israel Ltd. The rats were included in the study after an acclimatization period of at least five days. All animal experiments were carried out according to the United States National Institutes of Health/Department of Health and Human Services’ Guide for the Care and Use of Laboratory Animals (2011).
All animals were group-housed under climate-controlled conditions with a 12-hour light/dark cycle, a standard temperature of 20±3°C, and appropriate environmental enrichment (cardboard, tissues, red polycarbonate houses, and seeds) in the cages. Mice and rats had access to food and drinking water ad libitum.
Ponesimod was synthesized as previously described.39 The substance was stored at room temperature and protected from light. DMF was purchased from Sigma-Aldrich (St. Louis, Missouri). Both compounds had the same formulation: 0.5% methylcellulose (Sigma), 0.5% Tween 80 (Sigma) in water. Compounds and vehicle were given orally once or twice a day to animals at a volume of 10mL/kg in rats and 5 or 8mL/kg in mice at doses indicated in the legends of Figures 1–7.
Myelin oligodendrocyte glycoprotein (MOG)-induced EAE in C57BL/6 mice. Female C57BL/6 mice were immunized with 150micrograms of the encephalitogenic peptide MOG35-55 (MEVGWYRSPFSRVVHLYRNGK, from AusPepor GL Biochem), emulsified 1:1 (0.1mL: 0.1mL) in Complete Freund’s Adjuvant (CFA) (Difco) containing 4mg/mL Mycobacterium tuberculosis (BD). On Day 0, the mixture was injected subcutaneously into each flank of the abdomen. Immediately thereafter and again 48 hours later, each mouse was injected intravenously (IV) with 350ng of pertussis toxin (List Biological Laboratories) in 0.2mL of PBS. Ponesimod was administered orally at 30mg/kg twice a day in a preventative mode, starting from Day 1 after immunization, or in a therapeutic mode starting after onset of the disease (Day 15).
Mice were monitored daily, and the neurological impairment was quantified on an arbitrary 0-to-5 scale defined as 0=no detectable impairment; 1=flaccid tail; 2=hind limb weakness; 3=hind limb paralysis; 4=hind limb paralysis and ascending paralysis; and 5=deceased or euthanized. Following the recommendations of the animal ethics committee, mice were euthanized after reaching a clinical score of 4 on two consecutive days.
Histology. At the completion of the experiments, mice were euthanized and the brain and spinal cord were carefully removed and immersed in 10% buffered neutral formalin (Sigma) for seven days. Fixed tissues were dehydrated in ethanol gradient prior to their immersion in xylene (Grale) and embedment in paraffin wax. Sections of 5-micrometer thickness were cut on a microtome (Leica) from various regions of the spinal cord, cerebellum, and brain. Sections were stained with hematoxylin-eosin (HE), Luxol fast blue (LFB), and Bielschowsky for evidence of inflammation, demyelination, and axonal damage, respectively.
Semiquantitative evaluation of histological lesions. Semiquantitative histological evaluation for inflammation, demyelination, and axonal damage was performed and scored in a blinded fashion. From each mouse, 20 to 30 sections were examined. Inflammation scores were defined as follows: 0=no inflammation; 1=cellular infiltrates only in the perivascular areas and meninges; 2=mild cellular infiltrate in parenchyma; 3=moderate cellular infiltrate in parenchyma; and 4=severe and diffuse cellular infiltrate in parenchyma. Myelin breakdown was scored as follows: 0=no demyelination; 1=mild demyelination; 2=moderate demyelination; and 3=severe demyelination. Axonal damage was assessed as follows: 0=normal appearance; 1=axon loss less than 25 percent; 2=axon loss between 25 and 50 percent; 3=axon loss between 50 and 75 percent; and 4=axon loss greater than 75 percent.
Myelin basic protein (MBP)-induced EAE in Lewis rats. Female Lewis rats were immunized with 200µg myelin basic protein (MBP) (Sigma) in 100µl of phosphate buffer saline (PBS) emulsified in an equal volume of CFA (300µg of mycobacterium tuberculosis H37Ra). Briefly, on Day 0, rats were injected subcutaneously in the right paw and intradermally in the base of the tail 100µL of the MBP/CFA emulsion per injection site. Rats were randomized into treatment groups and orally administered ponesimod (30, 100mg/kg), DMF (40, 80, 120, 160mg/kg), or a combination of both compounds (100mg/kg ponesimod and 120mg/kg DMF) once daily from Day 0 until Day 21. Rats were monitored daily for body weight and signs of paralysis, which were scored according to a 0-to-15-point scale. The clinical score was determined by summing the score obtained for the tail, from 0=no signs; 1=half paralyzed tail; 2=fully paralyzed tail; and for each limb: from 0=no signs; 1=weak or altered gait; 2=paresis, 3=fully paralyzed limb; 15=dead animal. Blood samples were collected on Day 20, 24 hours after the last dosing of Day 19, and at euthanization on Day 21, one hour after the last dosing. Lymphocyte count was measured using ADVIA 120 blood analyzer (Siemens, Munich, Germany). No histological evaluation was performed at the end of the study because of the self-remitting nature of EAE disease in Lewis rats.
Statistical analysis. Statistical analysis was performed with GraphPad Prism software Version 7 (GraphPad Software, San Diego, California) using the tests specified in the legends of Figures 1–7. Results are expressed as mean+standard error of the mean (SEM).
Results
Ponesimod is effective both in a preventive and therapeutic setting in the MOG-induced EAE mouse model. The effect of preventative and therapeutic ponesimod on the clinical course of MOG-induced acute monophasic EAE was investigated in C57BL/6 mice. Vehicle-treated mice developed fulminant disease with severe clinical signs, which resulted in the death of all animals before Day 24, as indicated by a score of 5 (Figures 1A, 1B). Therapeutic treatment with ponesimod resulted in lower overall scores during the chronic phase, compared to vehicle, and a survival rate of 60 percent at the end of the study (Day 31). Preventive treatment resulted in a larger reduction of clinical scores and a 90-percent survival rate. (Figures 1A, 1B).
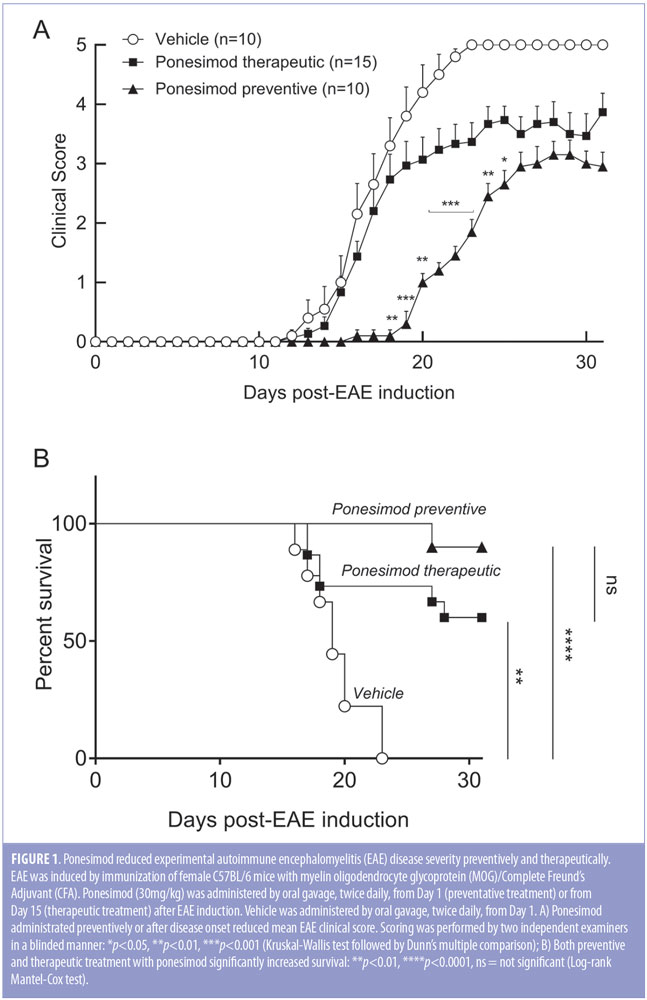
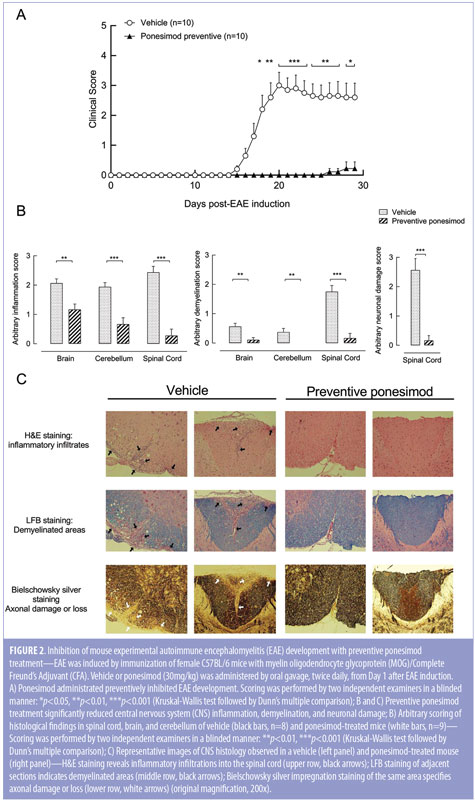
In a follow-up study with reduced overall disease severity (Figure 2A), the effect of ponesimod on cellular infiltration, demyelination, and axonal damage was evaluated in a preventive setting in the brain, spinal cord, and cerebellum (Figure 2B, 2C). In the vehicle-treated mice, onset of clinical signs was detected at Day 15 after immunization, reaching a maximal mean clinical score on Day 20. In contrast, mice treated preventively with ponesimod (30mg/kg twice daily from Day 1) were almost completely protected from the onset and progression of EAE. Only one out of the 10 mice treated with ponesimod started to develop minimal disease signs, which began manifesting on Day 26 (Figure 2A). Histological evaluation of the brain, cerebellum, and spinal cord confirmed the observed clinical scores in the respective groups. Compared to vehicle-treated mice, CNS tissues from ponesimod-treated mice revealed few if any inflammatory cell infiltrates, no signs of demyelination, and no apparent axonal damage or loss (Figures 2B, 2C). Prophylactic and therapeutic treatment with ponesimod did not reduce the titer of anti-MOG specific antibodies elicited following the immunization.
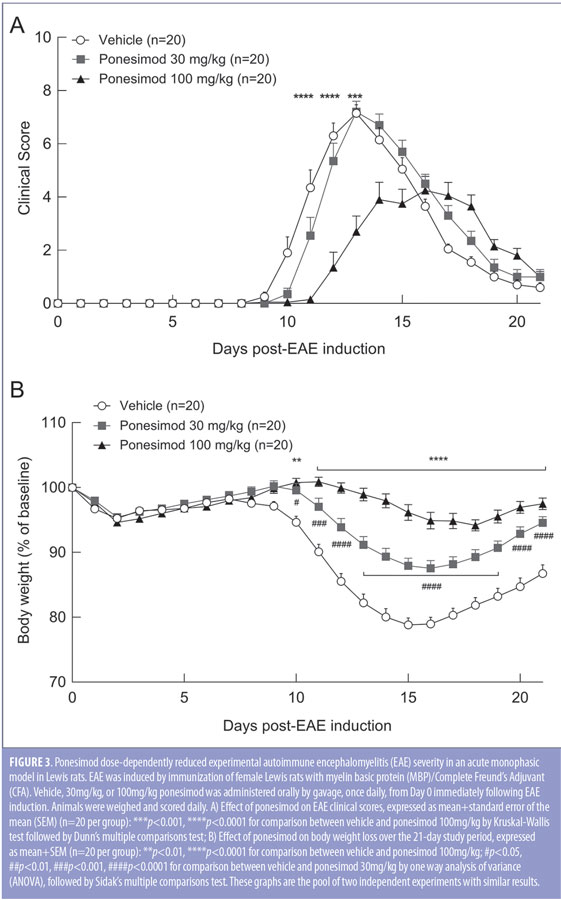
Ponesimod is effective in a rat EAE model. The dose-dependent efficacy of ponesimod was investigated in the acute monophasic EAE model in female Lewis rats. The vehicle-treated group developed first clinical signs at Day 9, reached the peak mean score at Day 13, followed by spontaneous remission. Preventive treatment with ponesimod at 30 and 100mg/kg delayed disease onset by one and two days, respectively. Only the highest dose of ponesimod significantly reduced the mean maximal clinical score (4.3±0.5, Day 16) in comparison with vehicle-treated group (7.2±0.3, Day 13) (Figure 3A). Furthermore, ponesimod treatment significantly reduced body weight loss in a dose-dependent manner, compared to vehicle-treated rats (Figure 3B).
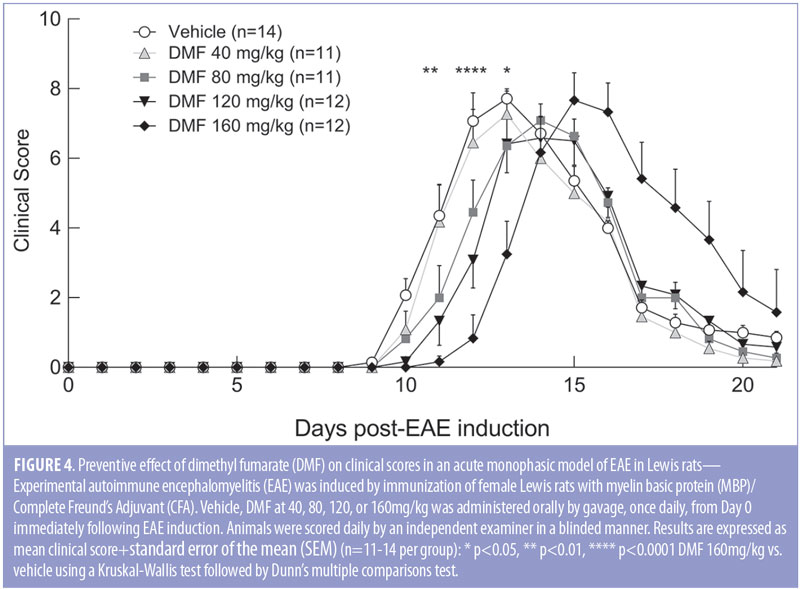
DMF slightly delays onset of disease in rat EAE. To define the optimal dose of DMF when combined with ponesimod in the acute monophasic EAE model using female Lewis rats, four doses of DMF were tested (40, 80, 120, and 160mg/kg). Preventive DMF treatment was given orally (by gavage) once daily, starting at Day 0 of immunization. The vehicle group developed first clinical signs at Day 9 and reached the peak score at Day 13 followed by characteristic remission. Treatment with DMF yielded a delay of disease onset by one (40, 80, and 120mg/kg) or two days (160mg/kg). Similarly, peak of disease was delayed by one (80 and 120mg/kg) or two days (160mg/kg). DMF reduced the clinical scores during the ascending phase of EAE in a dose-dependent manner; however, it did not reduce the severity and the overall extent of EAE (Figure 4). The highest dose of DMF was not tolerated, and one animal out of 12 died during the study.
Combined treatment of ponesimod with DMF is synergistic on clinical symptoms in a rat EAE model. Since the pharmacokinetic behavior of ponesimod in the rat is well described and highly reproducible,8 we used the Lewis rat model of EAE to study the effects of the combination of ponesimod with DMF. DMF at 120mg/kg was combined with ponesimod at 100mg/kg to test additive, or synergistic, clinical efficacy in the acute monophasic EAE rat model. Starting from the day of immunization, rats were treated orally (by gavage) with vehicle, ponesimod monotherapy (100mg/kg/day), DMF monotherapy (120mg/kg/day), or the combination of ponesimod with DMF. Combination therapy-treated rats exhibited significantly lower average clinical scores than vehicle- or monotherapy-treated rats. From Day 16 post-induction, the clinical score benefit of the combination was synergistic (Figure 5A). Only the combination treatment of ponesimod with DMF led to a full resolution of clinical signs at the end of the study period (Figure 5B). While DMF had no significant impact on the extent of the disease, administration of ponesimod monotherapy reduced the clinical score area under the curve by 77.6 percent, compared to vehicle-treated rats (Figure 5C).
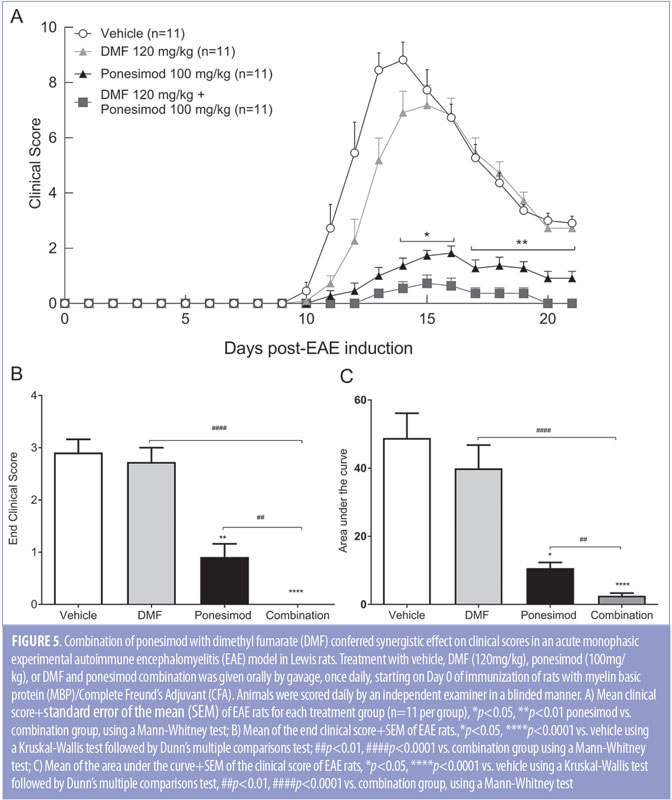
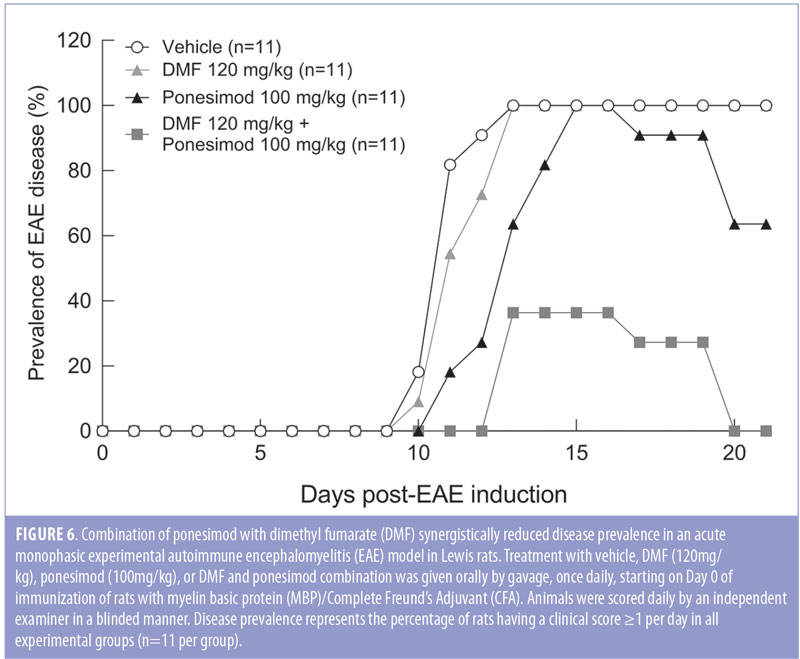
Clinical signs of EAE were evident (defined as a clinical score of at least 1 in 100 percent of the rats from Day 14 to the end of the study in vehicle and DMF-treated rats, and from Day 15 to Day 16 in ponesimod-treated rats. In contrast, combined treatment reduced the maximal disease prevalence to 36.4 percent (Day 13). Combination of ponesimod with DMF also accelerated the recovery, so that by Day 21, all the rats were symptom-free, compared to DMF and ponesimod monotherapy groups where 100 percent and 64 percent of the rats, respectively, had clinical signs (Figure 6).
Ponesimod induces the same blood lymphocyte count reduction in monotherapy and in combination with DMF in a rat EAE model. As previously shown in treatment-naïve rats,8 maximal lymphocyte count reduction also was achieved in EAE rats at 24 hours with 100mg/kg but not 30mg/kg ponesimod (data not shown). The repeated oral dosing of ponesimod resulted in a greater than 70-percent reduction of lymphocyte count one or 24 hours after the last administration. Combined treatment of ponesimod with DMF did not further reduce lymphocyte count since similar values were obtained in the ponesimod monotherapy group (Figure 7).

Discussion
In response to the rapid therapeutic progress in MS, treatment expectations have evolved, and the emerging goal for clinicians is to achieve NEDA in patients. MS is a complex disease, and the currently approved disease modifying therapies (DMTs), including the most aggressive MS treatments, have limited efficacy in preventing disability progression.4,40–42 In this context, combination therapies, which are the standard of practice in many chronic progressive diseases, are conceptually attractive. However, to date in clinical practice, there is no routinely used combination therapy in MS, and the largest and longest combination study in MS, the CombiRx study, which compared the combination of interferon beta-1a and glatiramer acetate to both agents alone, did not reveal a significant advantage of the combination.43 Nevertheless, experience with combination therapy in MS is still limited, and the availability of new oral treatments with different mechanisms of action provides opportunities to explore novel combination therapies for patients in whom monotherapy is only of limited value. Importantly, before starting such combination trials, any potential risk of negative interaction between the combination partners resulting in a reduced efficacy or aggravated disease must be minimized through preclinical PK/PD animal studies. Such combination trials must be carefully designed to select patient populations that will benefit most from the combination therapy without increased toxicity. Our study of the combination of ponesimod and DMF in MS models demonstrated for the first time its marked efficacy and its potential to reach NEDA. EAE is a well-recognized animal model for MS.38 All FDA-approved therapies for MS have shown efficacy in rodent EAE, giving a strong rationale to test the selective and rapidly reversible S1P1 modulator ponesimod and its combination with DMF using this model.
In the present study, ponesimod was effective preventively and therapeutically in reducing the overall clinical severity of the mouse EAE disease. Amelioration of EAE with ponesimod treatment was highlighted by improved histological outcomes. Ponesimod-treated mice showed reduced inflammatory cell infiltrates in the CNS, no signs of demyelination, and no apparent axonal damage or loss, compared to vehicle-treated mice. Ponesimod was further assessed in rats where the dose-dependent effect of ponesimod on total lymphocyte count has been well described8 and is highly reproducible among strains. The EAE model in the Lewis rat is the most commonly used MS model showing a monophasic disease course, with ascending and remitting phases, which mimic a relapse in RMS.44 Fingolimod was tested in this rat model and was shown to reduce both disease incidence and clinical score, most likely via inhibition of T-cell infiltration into the CNS.37 Similarly, preventative administration of ponesimod demonstrated a dose-dependent response on clinical outcomes. While the lowest dose had a moderate effect, mainly reducing body weight loss associated with EAE, the highest dose of ponesimod led to a 24-hour reduction of lymphocyte count and reduced the severity and the overall extent of the disease. Consistent with these preclinical EAE models, a large Phase II clinical trial evaluating safety and tolerability of ponesimod in patients with RMS was completed, demonstrating a dose-dependent therapeutic effect with acceptable safety and tolerability.45 Moreover, in a long-term extension of the Phase II study, ponesimod treatment showed a trend of dose-dependent decrease in disability accumulation and in brain volume loss.46 The evidence demonstrating the fast reversibility of lymphocyte count reduction and preservation of effector T cell function by ponesimod strongly support its effectiveness as an immunomodulatory drug for use in combination with another approved oral DMT. There has been considerable experience with DMF, an oral DMT approved in 2013 for RMS. Although the use of oral DMF yields significant benefit for the patient, it is only partially effective, with less than 25 percent of patients reaching NEDA.47 Nevertheless, the availability of partly effective therapeutics with mechanisms of action different than those of ponesimod provides the opportunity to target multiple factors of MS immunopathology when tested in combination, potentially leading to better suppression of disease activity.
Before evaluating the potential synergistic effect of combining ponesimod and DMF, we first performed a dose response of DMF in the EAE model in Lewis rats. Monotherapy of DMF at the maximal tolerated dose (120mg/kg) showed limited effect and was less effective than ponesimod, which might be due to the short-term treatment and the self-remitting nature of this rat EAE disease model. Indeed, the underlying complex mechanisms for the therapeutic efficacy of DMF, such as inhibition of the translocation of NF-kB and upregulation of cytoprotective genes, might need more time to achieve significant clinical benefit.
Combination of ponesimod and DMF showed synergistic effect and ameliorated EAE disease. Specifically, combination therapy delayed disease onset and reduced overall clinical scores and disease duration. Furthermore, close to 70 percent of the combination-treated rats were free from clinical signs during the entire study period, while 100 percent of the rats in the monotherapy-treated groups exhibited signs of paralysis at the acute phase of disease. Importantly, only the combination of ponesimod and DMF led to the full resolution of clinical signs at the end of the study. The combination treatment was well tolerated in rats, and there was no observed antagonistic interaction between DMF and ponesimod, as previously described with certain immunomodulatory agents, such as glatiramer acetate with Type I interferon.48
When considering the combination of different drugs, it is important to minimize risk of overlapping or increased toxicity. The sustained blood lymphocyte count reduction with ponesimod is a well-described pharmacological effect due to its mechanism of action.9 Several studies have shown that ponesimod, similar to fingolimod, has a predominant effect on naïve T cells and central memory CD4+ T cells, but spares effector T cells, especially CD8+ effector T cells.49,50 In contrast, DMF was shown to induce dose-dependent apoptosis of human T cells with a preferential effect on CD8+ T cells.51 However, lymphopenia develops in less than five percent of DMF-treated patients with MS, which is generally mild and appears after few months, reaching a nadir within 12 months.4,52 In the present study, DMF monotherapy did not induce lymphopenia, nor did the combined treatment further reduce the lymphocyte count. Compared to ponesimod alone, the synergistic effect observed on EAE severity in the combination group cannot be attributed to eliciting a stronger lymphocyte count reduction but rather through acting on different effector arms of EAE pathology. On the other hand, since in the present study DMF did not cause lymphopenia, it does not allow us to draw conclusions on the risk of combining both drugs regarding potential profound immunosuppression.
Conclusion
Our study demonstrated for the first time that ponesimod monotherapy is efficacious in mouse and rat EAE models, when combined with DMF, provides synergistic protection and complete resolution of disease in rat EAE. Both drugs have distinct immunomodulatory and neuroprotective properties that appear to act in a complementary manner, resulting in increased efficacy. While the precise mechanism and individual drug contribution in the combination setting is not fully understood, this combination has the potential to increase the proportion of patients achieving NEDA status in MS and supports the ongoing Phase III combination trial in MS (ClinicalTrial.gov Identifier: NCT02907177). Although both ponesimod and DMF can reduce lymphocyte count in humans, the mechanisms of action are different and are not expected to lead to a profound immunosuppression. In case of opportunistic infections or adverse events, ponesimod treatment can be safely interrupted while still administering DMF, and lymphocyte recovery can be expected within a few days, thus allowing for a safe combination.18,19 In addition, these encouraging preclinical data open the way to test ponesimod in combination with other oral drugs approved for the treatment of RMS, such as teriflunomide, where the different mechanisms of action of each agent might provide synergistic activity.
References
- Kim W, Zandona ME, Kim SH, Kim HJ. Oral disease-modifying therapies for multiple sclerosis. J Clin Neurol. 2015;11(1):9-19.
- Utz KS, Hoog J, Wentrup A, et al. Patient preferences for disease-modifying drugs in multiple sclerosis therapy: a choice-based conjoint analysis. Ther Adv Neurol Disord. 2014;7(6):263–275.
- Kappos L, Radue EW, O’Connor P, et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med. 2010;362(5):387–401.
- Gold R, Kappos L, Arnold DL, et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N Engl J Med. 2012;367(12):1098–1107.
- Oh J, O’Connor PW. Teriflunomide in the treatment of multiple sclerosis: current evidence and future prospects. Ther Adv Neurol Disord. 2014;7(5):239–252.
- D’Ambrosio D, Freedman MS, Prinz J. Ponesimod, a selective S1P1 receptor modulator: a potential treatment for multiple sclerosis and other immune-mediated diseases. Ther Adv Chronic Dis. 2016;7(1):18–33.
- Chun J, Hartung HP. Mechanism of action of oral fingolimod (FTY720) in multiple sclerosis. Clin Neuropharmacol. 2010;33(2):91–101.
- Piali L, Froidevaux S, Hess P, et al. The selective sphingosine 1-phosphate receptor 1 agonist ponesimod protects against lymphocyte-mediated tissue inflammation. J Pharmacol Exp Ther. 2011;337(2):547–556.
- Brinkmann V, Billich A, Baumruker T, et al. Fingolimod (FTY720): discovery and development of an oral drug to treat multiple sclerosis. Nat Rev Drug Discov. 2010;9(11):883–897.
- Pinschewer DD, Ochsenbein AF, Odermatt B, et al. FTY720 immunosuppression impairs effector T cell peripheral homing without affecting induction, expansion, and memory. J Immunol. 2000;164(11):5761–5770.
- Choi JW, Gardell SE, Herr DR, et al. FTY720 (fingolimod) efficacy in an animal model of multiple sclerosis requires astrocyte sphingosine 1-phosphate receptor 1 (S1P1) modulation. Proc Natl Acad Sci U S A. 2011;108(2):751–756.
- Pitteri M, Magliozzi R, Bajrami A, et al. Potential neuroprotective effect of Fingolimod in multiple sclerosis and its association with clinical variables. Expert Opin Pharmacother. 2018;19(4):387–395.
- Brinkmann V, Davis MD, Heise CE, et al. The immune modulator FTY720 targets sphingosine 1-phosphate receptors. J Biol Chem. 2002;277(24):21453–21457.
- Matloubian M, Lo CG, Cinamon G, et al. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature. 2004;427(6972):355–360.
- Murakami A, Takasugi H, Ohnuma S, et al. Sphingosine 1-phosphate (S1P) regulates vascular contraction via S1P3 receptor: investigation based on a new S1P3 receptor antagonist. Mol Pharmacol. 2010;77(4):704–713.
- Brinkmann V. Sphingosine 1-phosphate receptors in health and disease: mechanistic insights from gene deletion studies and reverse pharmacology. Pharmacol Ther. 2007;115(1):84–105.
- Camm J, Hla T, Bakshi R, Brinkmann V. Cardiac and vascular effects of fingolimod: mechanistic basis and clinical implications. Am Heart J. 2014;168(5):632–644.
- Jurcevic SHC, Greenlaw R, Juif PE, Dingemanse J. Effects of multiple-dose ponesimod, a selective S1P1 receptor modulator, on lymphocyte subsets in healthy humans. Drug Des Devel Ther. 2016;11:123–131.
- Juif PE, Kraehenbuehl S, Dingemanse J. Clinical pharmacology, efficacy, and safety aspects of sphingosine-1-phosphate receptor modulators. Expert Opin Drug Metab Toxicol. 2016;12(8):879–895.
- Schweckendiek W. [Treatment of psoriasis vulgaris]. Med Monatsschr. 1959;13(2):103–104.
- Gerdes S, Shakery K, Mrowietz U. Dimethylfumarate inhibits nuclear binding of nuclear factor kappaB but not of nuclear factor of activated T cells and CCAAT/enhancer binding protein beta in activated human T cells. Br J Dermatol. 2007;156(5):838–842.
- Treumer F, Zhu K, Glaser R, Mrowietz U. Dimethylfumarate is a potent inducer of apoptosis in human T cells. J Invest Dermatol. 2003;121(6):1383–1388.
- Loewe R, Holnthoner W, Groger M, et al. Dimethylfumarate inhibits TNF-induced nuclear entry of NF-kappa B/p65 in human endothelial cells. J Immunol. 2002;168(9):4781–4787.
- Gillard GO, Collette B, Anderson J, et al. DMF, but not other fumarates, inhibits NF-kappaB activity in vitro in an Nrf2-independent manner. J Neuroimmunol. 2015;283:74–85.
- Litjens NH, Rademaker M, Ravensbergen B, et al. Monomethylfumarate affects polarization of monocyte-derived dendritic cells resulting in down-regulated Th1 lymphocyte responses. Eur J Immunol. 2004;34(2):565–575.
- de Jong R, Bezemer AC, Zomerdijk TP, et al. Selective stimulation of T helper 2 cytokine responses by the anti-psoriasis agent monomethylfumarate. Eur J Immunol. 1996;26(9):2067–2074.
- Ghoreschi K, Bruck J, Kellerer C, et al. Fumarates improve psoriasis and multiple sclerosis by inducing type II dendritic cells. J Exp Med. 2011;208(11):2291–2303.
- Tahvili S, Zandieh B, Amirghofran Z. The effect of dimethyl fumarate on gene expression and the level of cytokines related to different T helper cell subsets in peripheral blood mononuclear cells of patients with psoriasis. Int J Dermatol. 2015;54(7):e254–260.
- Kornberg MD, Bhargava P, Kim PM, et al. Dimethyl fumarate targets GAPDH and aerobic glycolysis to modulate immunity. Science. 2018;360(6387):449–453.
- Albrecht P, Bouchachia I, Goebels N, et al. Effects of dimethyl fumarate on neuroprotection and immunomodulation. J Neuroinflammation. 2012;9:163.
- Dibbert S, Clement B, Skak-Nielsen T, et al. Detection of fumarate-glutathione adducts in the portal vein blood of rats: evidence for rapid dimethylfumarate metabolism. Arch Dermatol Res. 2013;305(5):447–451.
- Linker RA, Lee DH, Ryan S, et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain. 2011;134(Pt 3):678–692.
- Schilling S, Goelz S, Linker R, et al. Fumaric acid esters are effective in chronic experimental autoimmune encephalomyelitis and suppress macrophage infiltration. Clin Exp Immunol. 2006;145(1):101–107.
- Parodi B, Rossi S, Morando S, et al. Fumarates modulate microglia activation through a novel HCAR2 signaling pathway and rescue synaptic dysregulation in inflamed CNS. Acta Neuropathol. 2015;130(2):279–295.
- Merrill JE, Hanak S, Pu SF, et al. Teriflunomide reduces behavioral, electrophysiological, and histopathological deficits in the Dark Agouti rat model of experimental autoimmune encephalomyelitis. J Neurol. 2009;256(1):89–103.
- Duffy SS, Lees JG, Moalem-Taylor G. The contribution of immune and glial cell types in experimental autoimmune encephalomyelitis and multiple sclerosis. Mult Scler Int. 2014;2014:285245.
- Fujino M, Funeshima N, Kitazawa Y, et al. Amelioration of experimental autoimmune encephalomyelitis in Lewis rats by FTY720 treatment. J Pharmacol Exp Ther. 2003;305(1):70-77.
- Robinson AP, Harp CT, Noronha A, Miller SD. The experimental autoimmune encephalomyelitis (EAE) model of MS: utility for understanding disease pathophysiology and treatment. Handb Clin Neurol. 2014;122:173–189.
- Bolli MH, Abele S, Binkert C, et al. 2-imino-thiazolidin-4-one derivatives as potent, orally active S1P1 receptor agonists. J Med Chem. 2010;53(10):4198–4211.
- Cohen JA, Coles AJ, Arnold DL, et al. Alemtuzumab versus interferon beta 1a as first-line treatment for patients with relapsing-remitting multiple sclerosis: a randomised controlled phase 3 trial. Lancet. 2012;380(9856):1819–1828.
- Coles AJ, Twyman CL, Arnold DL, et al. Alemtuzumab for patients with relapsing multiple sclerosis after disease-modifying therapy: a randomised controlled phase 3 trial. Lancet. 2012;380(9856):1829–1839.
- Rudick RA, Stuart WH, Calabresi PA, et al. Natalizumab plus interferon beta-1a for relapsing multiple sclerosis. N Engl J Med. 2006;354(9):911–923.
- Lublin FD, Cofield SS, Cutter GR, et al. Long-term follow-up of a randomized study of combination interferon and glatiramer acetate in multiple sclerosis: efficacy and safety results up to 7 years. Mult Scler Relat Disord. 2017;18:95–102.
- Pitarokoili K, Ambrosius B, Gold R. Lewis rat model of experimental autoimmune encephalomyelitis. Curr Protoc Neurosci. 2017;81:9 61 61-69 61 20.
- Olsson T, Boster A, Fernandez O, et al. Oral ponesimod in relapsing-remitting multiple sclerosis: a randomised phase II trial. J Neurol Neurosurg Psychiatry. 2014;85(11):1198–1208.
- Pozzilli CFO, Olsson T, Freedman MS, et al. Maintenance of efficacy, safety and tolerability of ponesimod in patients with relapsing remitting multiple sclerosis: phase II extension study. Poster presented at ECTRIMS 2013.
- Nixon R, Bergvall N, Tomic D, et al. No evidence of disease activity: indirect comparisons of oral therapies for the treatment of relapsing-remitting multiple sclerosis. Adv Ther. 2014;31(11):1134–1154.
- Brod SA, Lindsey JW, Wolinsky JS. Combination therapy with glatiramer acetate (copolymer-1) and a type I interferon (IFN-alpha) does not improve experimental autoimmune encephalomyelitis. Ann Neurol. 2000;47(1):127–131.
- Mehling M, Johnson TA, Antel J, et al. Clinical immunology of the sphingosine 1-phosphate receptor modulator fingolimod (FTY720) in multiple sclerosis. Neurology. 2011;76(8 Suppl 3):S20–27.
- D’Ambrosio D, Steinmann J, Brossard P, Dingemanse J. Differential effects of ponesimod, a selective S1P1 receptor modulator, on blood-circulating human T cell subpopulations. Immunopharmacol Immunotoxicol. 2015;37(1):103–109.
- Ghadiri M, Rezk A, Li R, et al. Dimethyl fumarate-induced lymphopenia in MS due to differential T-cell subset apoptosis. Neurol Neuroimmunol Neuroinflamm. 2017;4(3):e340.
- Fox RJ, Miller DH, Phillips JT, et al. Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. N Engl J Med. 2012;367(12):1087–1097.