 by Amanda K. Kitten, PharmD; Sarah A. Hallowell, PharmD; Stephen Saklad, PharmD, BCPP; and Kirk E. Evoy, PharmD, BCACP, BC-ADM, CTTS
by Amanda K. Kitten, PharmD; Sarah A. Hallowell, PharmD; Stephen Saklad, PharmD, BCPP; and Kirk E. Evoy, PharmD, BCACP, BC-ADM, CTTS
Drs. Kitten, Hallowell, Saklad, and Evoy are with the College of Pharmacy, The University of Texas at Austin in Austin, Texas.
Innov Clin Neurosci. 2017;15(1–2):16–22
FUNDING: The authors received no funding for the development of this manuscript.
DISCLOSURES: Dr. Saklad declares the following: employee of The University of Texas at Austin College of Pharmacy; appointed to the Texas Department of State Health Services, San Antonio State Hospital and the UT Health Science Center San Antonio School of Medicine; speakers bureau for Otsuka / Lundbeck; consultant for NCS Pearson, and Takeda; speaker for several professional organizations; Board of Directors for the College of Psychiatric and Neurologic Pharmacists Foundation; Business Development Council for the College of Psychiatric and Neurologic Pharmacists; expert witness on both defendant and plaintiff sides; no direct stock ownership in pharmaceutical corporations. Drs. Kitten, Hallowell, and Evoy have no conflicts of interest relevant to the content of this article.
ABSTRACT: Objective: Pimavanserin is the first United States Food and Drug Administration (FDA)-approved treatment for Parkinson’s disease psychosis (PDP). This article reviews the safety, efficacy, and pharmacology data for pimavanserin and its role in therapy. Method of Research: Initial literature sources were identified via MEDLINE search (1946–September 2016) of pimavanserin and ACP-103 (original molecular designation). Reference review and search of FDA.gov and clinicaltrials.gov yielded additional studies. English-language studies of pimavanserin for PDP were evaluated. Animal studies were excluded. Randomized, controlled trials (RCTs) were prioritized. Results: Four RCTs were identified. In each, pimavanserin was well-tolerated with few adverse effects and no worsening of motor symptoms. A Phase II trial displayed a nonsignificant trend toward Scale for Assessment of Positive Symptoms (SAPS) improvement (p=0.09), with significant benefits in secondary efficacy markers. However, two Phase III trials, including one that was terminated early, failed to show significant SAPS improvement. A third Phase III trial with an improved research design utilized a nine-item subset of the SAPS, the SAPS-PD, as the primary outcome and demonstrated that pimavanserin 40mg was effective in improving PDP compared to placebo (p=0.0014, effect size=0.50). Secondary outcomes were also significantly improved: Clinical Global Impression of Severity (CGI-S) (p=0.0007, effect size=0.52) and Clinical Global Impression of Improvement (CGI-I) (p=0.0011, effect size=0.51), caregiver burden (p=0.0016, effect size=0.50), nighttime sleep (p=0.0446, effect size=0.31), and daytime wakefulness (p=0.012, effect size=0.39). Conclusion: Evidence suggests pimavanserin attenuates PDP symptoms with few adverse effects and little risk of worsening motor function. With limited treatment options for PDP, pimavanserin represents an important therapeutic innovation.
KEYWORDS: Pimavanserin, ACP-103, Parkinson’s disease psychosis
INTRODUCTION
Parkinson’s disease (PD) is a prevalent disorder that affects approximately 40 to 1,000 persons/100,000, with increased occurrence in older individuals.[1] PD is characterized by progressive neurodegeneration that manifests in the early stages as mild cognitive impairment and moderate motor dysfunction, namely bradykinesia, rigidity, and tremor.[2,3] As PD advances, the severity escalates, culminating in significant motor dysfunction, such as freezing of gait, falls, and choking, and neuropsychiatric complications, including psychosis. PD psychosis (PDP) occurs in up to 75 percent of PD cases and is associated with increased caregiver burden, nursing home admission, and mortality.[2–5] Manifestations of PDP include visual hallucinations (VH), the most common presentation, which is observed in up to 70 percent of patients with PDP. Other presentations of PDP include delusions, illusions, and auditory or presence hallucinations.[6]
Despite the need to reduce morbidity and mortality associated with PDP, treatment options are limited. Psychosis development might be partially attributable to medications used to treat PD motor dysfunction.7 As a result, first-line therapy for treating PDP consists of reducing or discontinuing anticholinergic medications, monoamine oxidase inhibitors (MAOI), levodopa, or dopamine agonists (DA), the cessation of which might worsen motor symptoms.[6,8] However, these medications are likely not the sole factor in the development of PDP, as symptoms tend not to directly correlate with medication usage, and patients might continue to experience PDP after these drugs are discontinued.[9]
If medication adjustment is not appropriate or fails to resolve symptoms, second-generation antipsychotics may be prescribed for PDP.[10,11] These medications are not approved by the United States Food and Drug Administration (FDA) for the treatment of PDP, and among the antipsychotics, only clozapine has compelling evidence to support its effectiveness in PDP.
Given the morbidity, mortality, and societal costs associated with PDP and dearth of effective, well-tolerated medications, new treatment options are needed.[10,11] Pimavanserin (NuplazidTM, Acadia Pharmaceuticals®, San Diego, California) is an antipsychotic with a unique mechanism of action that functions as a highly selective, partial inverse agonist at 5-HT2A receptors.[12,13] Pimavanserin shows promise as a safe, effective addition to the PDP armamentarium and in 2016 became the first drug to receive FDA approval for PDP treatment.[14] This article reviews the pharmacology, efficacy, and safety data available for pimavanserin and provides comparison with other therapeutic options to suggest its likely role in treatment of PDP.
METHODS
Initial literature sources were identified via MEDLINE search (1946–September 2016) of pimavanserin and ACP-103 (the original molecular designation of pimavanserin). Reference review and search of FDA and Acadia® websites and clinicaltrials.gov yielded additional unpublished data. English-language studies of pimavanserin for PDP were evaluated. Animal studies were excluded. Priority was given to randomized, controlled trials (RCTs).
Abstracts of identified studies were screened for eligibility, initially by two reviewers, with 100-percent agreement. A third reviewer assessed the included studies post-agreement by the initial reviewers. Details of the trials were extracted into a spreadsheet. The data were summarized with regard to the drug’s pharmacology, pharmacokinetics, clinical trial data, dosing, adverse effects, precautions, contraindications, monitoring, and drug interactions.
RESULTS
Pharmacology. The primary pathology in PD is dopamine deficiency in the basal ganglia, which results in the characteristic motor dysfunction.[7] Dopaminergic neuronal degeneration in the nigrostriatal pathway produces overall increased inhibitory signaling to the thalamus and motor cortex, thus resulting in bradykinesia, muscular rigidity, resting tremor, and postural instability.[15] Though development of PDP likely involves multiple systems, including dopaminergic, glutamatergic, cholinergic, and serotonergic systems, some researchers have hypothesized that dopamine deficiency precipitates up-regulation of 5-HT function and receptor sensitivity, especially in the visual processing circuitry. This can result in psychosis with prominent VH, most commonly with insight.[7,9,13,16,17] Of particular interest are 5-HT2A receptors, as brain imaging scans of patients with PD with VH display increased 5-HT2A binding in the brain’s visual processing areas.[17,18] Medications currently used off-label to treat PDP display prominent 5-HT2 receptor binding at low doses, consistent with this theory.[7,16]
Pimavanserin primarily functions as an inverse agonist and antagonist (a partial inverse agonist) at 5-HT2A receptors.[12] Selectivity for 5-HT2 receptors and sparing the dopamine post-synaptic receptors differentiates pimavanserin from other antipsychotic drugs currently used in PDP.[16] Pimavanserin binds with high affinity (Ki 0.087nM) to 5-HT2A and five-fold lower affinity (Ki 0.44nM) to 5-HT2C, with negligible binding at 5-HT2B, dopaminergic (D3), muscarinic (M5), and opioid (sigma 1) receptors.[19] First-generation antipsychotics should be avoided in PDP, as their extensive dopamine (D2) receptor antagonism worsens motor dysfunction.[7] Second-generation antipsychotics antagonize 5-HT2A receptors in addition to D2 receptors, but motor dysfunction might still be encountered. However, the relative amount of 5-HT2 and D2 antagonism is highly variable among second-generation antipsychotics. Those with very low D2 affinity, such as clozapine and quetiapine, are currently the most commonly used medications to treat PDP.[7,10,11,16]
Pharmacokinetics. Following administration of single oral doses of pimavanserin, brain imaging scans display dose-proportional pharmacokinetics.[12] Pimavanserin has one major active metabolite, AC-279. Mean plasma half-life of pimavanserin is 57 hours, and AC-279 is 200 hours. Pimavanserin’s median time to peak concentration (Tmax) is six hours (range 4–24), irrespective of dose. Median Tmax of the formation of AC-279 is also six hours. Mean maximum concentration (Cmax) after a single, 100mg oral dose of pimavanserin in a fasted state is 57mg/mL, whereas mean area under the curve (AUC) under the same conditions is 3871h*ng/mL.[20] Pimavanserin tablets are 99.7-percent bioavailable.[21] Neither rate nor extent of pimavanserin exposure is significantly affected by concurrent ingestion of a high-fat meal, as Cmax decreases nine percent while AUC increases eight percent.[16,20,21] Average volume of distribution for pimavanserin following a single dose is 2,173L.[12] Pimavanserin is 95-percent protein bound in human plasma.
Metabolism occurs primarily via cytochrome P450 enzyme (CYP) 3A4, the major contributor to formation of the active N-desmethylated metabolite, as well as CYP3A5.[12] To a lesser degree, pimavanserin is also metabolized via CYP2J2, CYP2D6, and other CYP and flavin-containing monooxygenase (FMO) enzymes. However, pimavanserin and metabolites do not inhibit or induce CYP enzymes to a clinically significant degree. Transporters do not appear to play a role in pimavanserin disposition. After an oral dose, 0.55 percent was excreted unchanged in the urine, while 1.53 percent was eliminated in feces.
Pimavanserin pharmacokinetics are not significantly affected by weight, age, sex, or ethnicity.[12] Exposure in patients with mild-to-moderate renal impairment is similar to that of patients with normal renal function. Pharmacokinetic studies in patients with hepatic impairment or severe renal dysfunction are currently underway.[22]
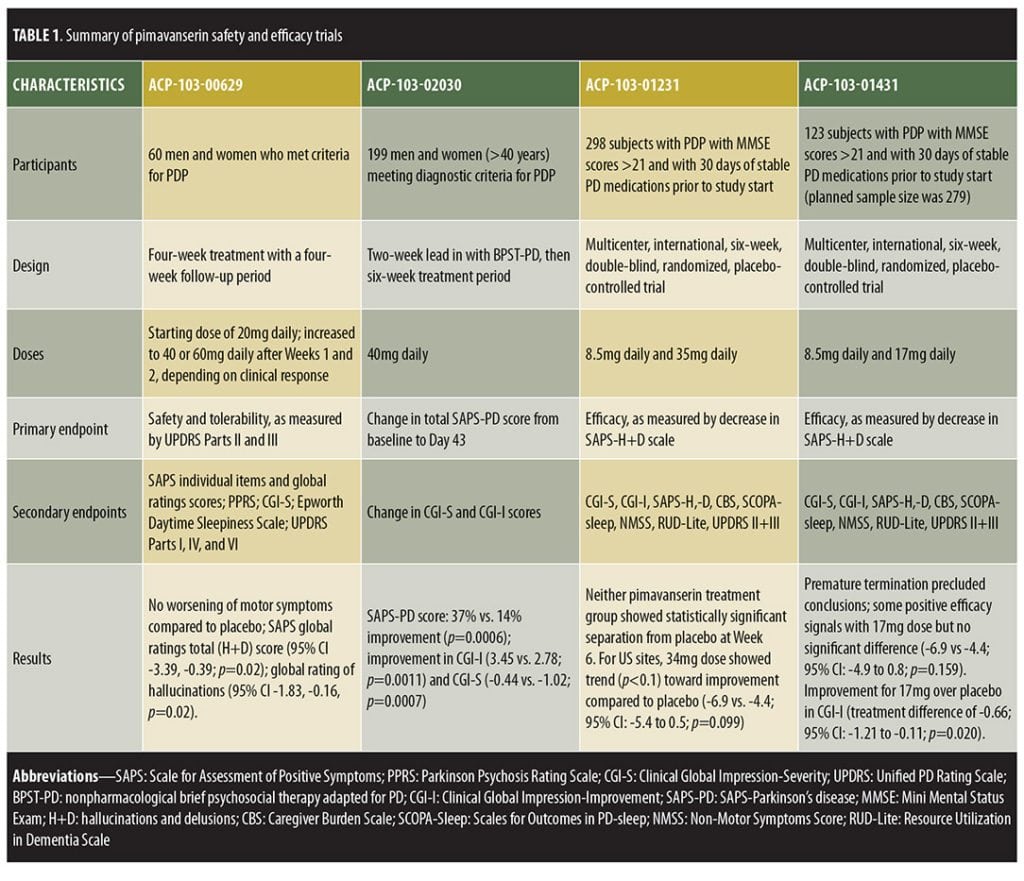
Clinical trials. Four RCTs have assessed pimavanserin in PDP, each showing that pimavanserin is safe and without negative impact on PD motor symptoms.[22–24] Preliminary data from two ongoing Phase IV, open-label, extension studies further support the relative safety of pimavanserin.[22] With regard to efficacy, only one of the four trials exhibited statistically significant improvement in the primary efficacy endpoint.[22–24] Among the earlier trials that did not demonstrate statistical significance, a Phase II study (ACP-103-006) was completed with results published that displayed a nonsignificant trend toward improvement in the primary outcome, along with significant improvement in a number of secondary efficacy measures.[23] Two other trials failed to show efficacy and are currently unpublished (ACP-103-012 and ACP-103-014), though data from these studies are available via the FDA and meeting abstracts.[22,25–27] Despite these early studies failing to show statistical significance, the FDA agreed to review the New Drug Application (NDA) submission of pimavanserin following the aforementioned single trial displaying significant improvement in PDP (ACP-103-020).
ACP-103-006, a Phase II study powered to detect effects on motor symptoms through the United Parkinson’s Disease Rating Scale (UPDRS) Parts II and III, enrolled 60 subjects in the United States (US) with moderate-to-severe PDP. Subjects were randomly assigned to pimavanserin or placebo in a 1:1 ratio for four weeks, showing the drug to be well-tolerated and effective in some, but not all, efficacy measures.[22,23] Pimavanserin tartrate was initiated at 20mg with optional titration to 40mg or 60mg based on clinical response (mean dose standard deviation [SD]=44.8±16mg). Instead of worsening motor symptoms, a nonsignificant improvement was observed with both pimavanserin (-3.05, 95% confidence interval [CI]=-6.56-0.46) and placebo (-3.86, 95% CI= -7.20-0.53) in UPDRS II and III, with no difference between groups (p=0.74). The primary efficacy measure was the Scale for the Assessment of Positive Symptoms (SAPS) total score, which trended toward greater improvement with pimavanserin, though statistical significance was not reached (p=0.09). However, pimavanserin did produce significant improvement in the SAPS global ratings of hallucinations (p=0.02, effect size=0.58) and delusions (p=0.03, effect size=0.53), with trends toward improvement in SAPS total hallucination (p=0.16) and total delusion (p=0.06) domain scores. Additional measures of psychosis, including the Parkinson’s Psychosis Rating Scale (PPRS), Clinical Global Impression (CGI), and Epworth Daytime Sleepiness Scale, also displayed nonsignificant trends toward improvement in the pimavanserin group.
ACP-103-012, a randomized, double-blind, placebo-controlled trial (N=298) investigated pimavanserin tartrate at doses of 10mg and 40mg and enrolled subjects from the US, five European countries, and India.[22,25,26] Efficacy was not statistically different between the placebo and pimavanserin arms (SAP-hallucinations + delusions [H+D] least-squares [LS] mean difference from baseline versus placebo for 10mg [-0.07, 95% CI -1.7-1.59] and 40mg [-1.16, 95% CI -2.83-0.51]), which the investigators attributed to an enhanced placebo response.[22] ACP-103-014 was conducted concurrently in the US, eight European countries, and India, but used lower doses (10mg and 20mg). This study was terminated early, as investigators deemed the trial unlikely to display efficacy due to lack of significant improvement in ACP-103-012.
Several design changes were incorporated to improve the ability to detect a treatment difference in the pivotal Phase III trial (ACP-103-020).[24,28] ACP-103-020 was developed by identifying factors in ACP-103-012 and ACP-103-014 that positively correlated with effect size and applying those factors to the study design of ACP-103-020.[22] Thus, the investigators altered their methods, performing the study only in the US and Canada, incorporating centralized raters to reduce inter-rater variability, exclusively using the 40mg dose, employing a two-week lead-in period of psychosocial therapy to blunt the placebo response, and measuring a different primary outcome.[24] Whereas the three previous trials had used the SAPS total score as the primary outcome, ACP-103-020 employed a novel scale, the nine-item subset SAPS-PD.[23–25] While SAPS was originally developed for schizophrenia, SAPS-PD represents a more tailored scale, measuring only the symptoms of SAPS most relevant to PDP, based on symptom frequency from previous PDP studies.[21] In a study conducted to validate the SAPS-PD measure, a clinically significant change was defined as 2.33 points equivalent to a one-point change in the CGI-improvement scale (CGI-I).[29]
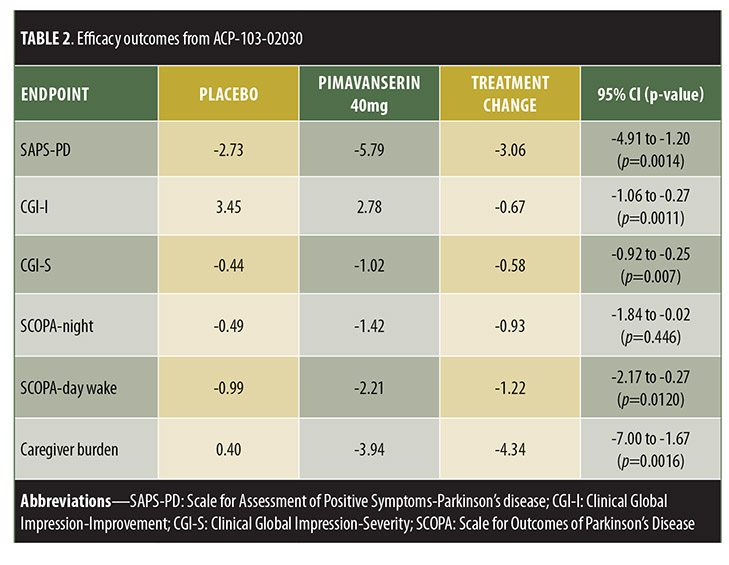
ACP-103-020, a six-week, double-blind, placebo-controlled trial, randomized 199 patients to placebo or pimavanserin tartrate 40mg.[24] Inclusion criteria included being at least 40 years old with psychotic symptoms that occurred at least weekly, were severe enough to warrant treatment, and were present for at least one month prior to enrollment. Those treated with pimavanserin exhibited significant improvement in SAPS-PD compared to placebo (-5.79 [37% improvement] vs. -2.73 [14% improvement], respectively, p=0.0006). Pimavanserin-treated patients also displayed significantly greater improvement in other measures of psychosis, including the full 20-item SAPS-H+D score (pimavanserin [-6.51] vs. placebo [-3.14], p=0.0012), CGI-I (effect size 0.51, p=0.0011), and CGI-Severity (CGI-S, effect size=0.52, p=0.0007) scores. Thus, this trial showed a statistically significant decrease in SAPS-H+D, which was measured as the primary outcome in previous trials, indicating that the positive results from this trial were not simply a result of using the new SAPS-PD measurement as the primary outcome. Exploratory analyses displayed significant improvement versus placebo in caregiver burden (effect size=0.50, p=0.0016), as well as nighttime sleep (effect size=0.31, p=0.0446) and daytime wakefulness (effect size 0.39, p=0.0120) according to the Scale for Outcomes in Parkinson’s Disease-Sleep Scale (SCOPA-Sleep). Both groups demonstrated nonsignificant motor symptom improvements, with no between-group difference (95% CI -2.14-2.72).
Currently completed studies assessed six weeks of pimavanserin treatment.[24] To evaluate long-term safety and efficacy in larger patient populations, two ongoing open-label extension studies are underway.[25] While one extension trial is studying the currently approved dose of 34mg, the other incorporates flexible dosing up to 51mg. Preliminary data continue to demonstrate efficacy and safety of pimavanserin, providing early support for the long-term and early use of pimavanserin in PDP.[22,27] Additional studies to ascertain the effects of hepatic and renal failure on pimavanserin’s tolerability and pharmacokinetics are also being conducted, as is a Phase II study of pimavanserin for the treatment of Alzheimer’s disease psychosis.[22] A completed analysis of pimavanserin for schizophrenia showed safety and efficacy when used with risperidone.[30]
Trial designs are summarized in Table 1 and results in Table 2.
Dosing. Pimavanserin is available as 17mg tablets.[12] FDA-approved dosing for PDP-associated hallucinations and delusions is 34mg once daily (equivalent to 40mg pimavanserin tartrate used in ACP-103-020), administered orally as two 17mg tablets with no dose titration. It can be taken with or without food.
Manufacturer labeling recommends considering dose adjustments when prescribed concomitantly with CYP3A4 inhibitors or inducers.[12] When co-administered with strong CYP3A4 inhibitors (e.g., ketoconazole), the recommended dose is 17mg daily. Patients taking strong 3A4 inducers (e.g., rifampin) should be monitored for reduced efficacy. A dose increase might be necessary in such cases, though specific recommendations are not provided.
Adverse effects. Pimavanserin appeared relatively safe and well-tolerated in pre-marketing clinical trials, with adverse events (AEs) occurring at rates similar to placebo.[23–25] In both the pivotal study (ACP-103-020) and the pooled clinical trial data presented in the pimavanserin prescribing information, peripheral edema (pooled data: 7% vs. 2%) and confusional state (pooled data: 6% vs. 3%) were the only AEs occurring in at least five percent of patients and at least twice the rate of placebo.[12,24] Importantly, worsening of motor symptoms has not been observed.
An additional concern with pimavanserin therapy is possible QT prolongation, which occurred in 19.5 percent of patients.[25] ACP-103-020 displayed a mean 7.3ms QTcB (Bazett’s corrected formula) interval increase with pimavanserin.[24] Sporadic QTcF (Frederica’s corrected formula) values of 500ms or greater and changes from baseline 60ms or greater have been reported with 34mg pimavanserin.[25] Compared with placebo, increased rates of ventricular repolarization-related AEs, such as torsades de pointes, have not yet been reported.
Precautions, contraindications, drug monitoring. As with all antipsychotic drugs, pimavanserin labeling includes a boxed warning for increased mortality in elderly patients with dementia-related psychosis, as previous studies of antipsychotic drugs have detected an increased risk of all-cause mortality in this population.[12] Preliminary data from ongoing extension studies revealed five deaths (pimavanserin [4] vs. placebo [1], estimated odds ratio [OR] 2.94, p=0.61) that were attributed to probable myocardial infarction, septic shock, septicemia, or respiratory distress among those in the pimavanserin group.[25] Clinicians should exercise caution before prescribing pimavanserin to patients at risk for QT prolongation, and should avoid prescribing pimavanserin altogether to patients with known QT prolongation.[16]
Drug interactions. As pimavanserin is metabolized by CYP3A4, healthcare providers should be aware of potential drug interactions.[12]1 Concomitant use of pimavanserin with strong CYP3A4 inhibitors (e.g., ketoconazole) or inducers (e.g., rifampin) warrants dose reduction or careful monitoring for efficacy, respectively. Furthermore, clinicians should exercise caution when prescribing pimavanserin concurrently with other drugs that cause prolonged QTc intervals, including class 1A antiarrhythmics (e.g., amiodarone), antipsychotics (e.g., ziprasidone), and fluoroquinolones (e.g., moxifloxacin).
DISCUSSION
Considerations for therapy. Given the limited number of effective, well-tolerated treatments for PDP, novel options are clearly needed. Pimavanserin represents the first FDA-approved PDP medication.[14] One clinical trial has shown pimavanserin to be efficacious, while multiple trials have demonstrated that it is well-tolerated with few side effects and no worsening of PD motor function.[22–24] Although two Phase III clinical trials failed to show improvement in the primary efficacy endpoint, the subsequent Phase III trial was successful after the investigators optimized the trial design to blunt placebo effect and implemented a primary outcome measure, the SAPS-PD, that was more sensitive to PDP improvement.[31] Furthermore, pimavanserin significantly improved sleep-wake cycle and decreased caregiver burden, suggesting enhanced quality of life (QOL) beyond attenuation of psychotic symptoms.[24] Reduced caregiver burden is particularly significant, as this could potentially delay the need for skilled nursing facility placement.
Current guidelines recommend cessation or gradual withdrawal of antiparkinsonian medications, if tolerated, as the first-line therapy for PDP.[8] In many cases, this is impossible, as discontinuation worsens PD motor symptoms. For severe psychosis refractory to such medication cessation or in patients unable to tolerate cessation due to worsened motor function, certain second-generation antipsychotics are most commonly used.[10,11] Their efficacy in PDP is likely attributed to effects as 5HT2A receptor antagonists.[16] In contrast to pimavanserin’s 5HT2A selectivity, second-generation antipsychotics also antagonize dopamine receptors and might worsen motor symptoms.
Clozapine, quetiapine, and olanzapine, second generation antipsychotics studied in PDP due to their relative lack of extrapyramidal symptoms, were found to be effective, possibly effective, and not effective, respectively. Most recent guidelines endorse the use of clozapine (Level B), with a less strong recommendation for quetiapine (Level C).[10,11] Neither clozapine nor quetiapine tend to significantly worsen motor dysfunction.[32,37] However, these drugs are not without limitations.
Clozapine can cause agranulocytosis, requiring frequent monitoring, and has a greater risk of other AEs, such as anticholinergic effects, hypotension, sedation, and metabolic abnormalities. The risk for agranulocytosis in particular necessitates that healthcare providers and pharmacists participate in a laborious risk evaluation and mitigation strategy (REMS) to prescribe or dispense clozapine.[10–14,38] These REMS requirements include frequent lab monitoring and documentation, which reduces clozapine’s utility and results in underutilization.[39] Despite its common use due to greater tolerability relative to clozapine, clinical trial data regarding quetiapine efficacy are weak, as most studies did not display significant improvements in psychosis.[35–37] While clozapine and quetiapine display a low propensity to significantly worsen PD motor symptoms, many antipsychotics, especially first-generation antipsychotics, have higher risk of motor-related AEs that limits their usefulness in the treatment of PDP.[16] Conversely, pimavanserin appears well-tolerated, and preliminary data from ongoing extension studies show pimavanserin to be safe and effective when taken for up to eight years, with total exposure of pimavanserin in PDP exceeding 825 patient years.[25–26] These data suggest promising long-term safety with pimavanserin. Finally, the relative tolerability of pimavanserin might allow the drug to be used earlier in the disease process, whereas clozapine treatment is typically delayed, which results in initiation of medication after the point at which PDP has already caused a noteworthy decline in the patient’s QOL.[40]
Multiple trials have demonstrated clozapine’s efficacy in PDP, and a head-to-head study showed clozapine to be more effective than quetiapine.[32–34,40,41] No head-to-head trials between pimavanserin and comparator drugs have been conducted. Comparing the pimavanserin trial data to previous clozapine trials, clozapine demonstrated a greater effect size compared to pimavanserin based on CGI-S, which was the only common efficacy outcome among key studies.[24,32] However, the clozapine trials were shorter and included fewer patients with more advanced psychosis, limiting comparisons of this data.[23,24,32,41]
Despite these advantages, price might be prohibitive, as pimavanserin costs $80/day (drug acquisition cost) for the recommended 34mg dose, whereas clozapine (exclusive of monitoring costs) and quetiapine cost $1.25 and $6.86 per day, respectively.[42,43] It is likely that many insurance companies covering the medication will restrict its use, require prior authorization for coverage, or include the medication in a high co-pay tier, given its cost.
Patients with PD should be routinely monitored for the development of PDP. If PDP is detected, careful discussion with the patient and caregivers should include the possible treatment options, beginning with reductions or adjustments in PD medications. Pimavanserin appears to be a better-tolerated but possibly less efficacious treatment for PDP than clozapine. In patients unable to obtain relief with antiparkinsonian medication dose-reduction alone, it is reasonable to consider a trial of pimavanserin if the patient and provider agree that the benefits of better tolerability outweigh the higher cost. If cost is a significant obstacle or pimavanserin fails, then they might consider a trial of clozapine.
SUMMARY
Though early trials failed to show significant improvement in the primary efficacy outcome, after adjustment in trial design, particularly to blunt placebo response and measure a primary outcome more specific to symptoms associated with PDP, the pivotal Phase III study ACP-103-020 showed pimavanserin effective in improving PD psychosis. Throughout the clinical trial program, pimavanserin appeared safe and well-tolerated, particularly with respect to its lack of deleterious effects on motor function.
Given that pimavanserin boasts the first FDA-approved indication for treatment of PDP, has the most data in well-conducted trials of PDP patients, and appears safer and better tolerated than current alternatives, pimavanserin is likely to assume a significant role in the treatment of PDP. While past studies of clozapine indicate it might provide similar or greater improvement in PDP, pimavanserin is likely to produce fewer AEs and require less monitoring. Future prospective studies directly comparing pimavanserin to alternative therapies are needed to identify the most ideal treatment for PDP.
REFERENCES
- Pringsheim T, Jette N, Frolkis A, Steeves TDL. The prevalence of Parkinson’s disease: a systematic review and meta-analysis. Mov Disord. 2014;29(13):1583–90.
- Martinez-Martin P, Rodriguez-blazquez C, Forjaz MJ, et al. Neuropsychiatric symptoms and caregiver’s burden in Parkinson’s disease. Parkinsonism Relat Disord. 2015;21(6):629–34.
- Kalia LV, Lang AE. Parkinson’s disease. Lancet. 2015;386:896–912.
- Goetz CG, Stebbins GT. Risk factors for nursing home placement in advanced Parkinson’s disease. Neurology. 1993;(43)2227–2229.
- Aarsland D, Larsen JP, Tandberg E, Laake K. Predictors of nursing home placement in Parkinson’s disease: a population-based, prospective study. JAGS. 2000;48:938–942.
- Cooney JW, Stacy M. Neuropsychiatric Issues in Parkinson’s disease. Curr Neurol Neurosci Rep. 2016;16(5):49.
- Chang A, Fox SH. Psychosis in Parkinson’s disease: epidemiology, pathophysiology, and management. Drugs. 2016;76(11):1093–1118.
- Patel T, Chang F. Parkinson’s disease guidelines for pharmacists. Can Pharm J (Ott). 2014;147(3):161–170.
- Ravina B, Marder K, Fernandez HH. Diagnostic criteria for psychosis in Parkinson’s disease: report of an NINDS, NIMH work group. Movement Disord. 2007;22(8):1061–1068.
- Miyasaki JM, Shannon K, Voon V, et al. Practice parameter: evaluation and treatment of depression, psychosis, and dementia in Parkinson’s disease (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2006;66:996–1002.
- Seppi K, Weintraub D, Coelho M, et al. The Movement Disorder Society Evidence-based Medicine Review Update: treatments for the non-motor symptoms of Parkinson’s disease. Mov Disord. 2011;26(3 suppl):S42–80.
- Nuplazid (pimavanserin) [package insert]. San Diego, CA: Acadia Pharmaceuticals Inc.; 2016.
- Hacksell U, Burstein ES, Mcfarland K, et al. On the discovery and development of pimavanserin: a novel drug candidate for Parkinson’s psychosis. Neurochem Res. 2014;39(10):2008–2017.
- United States Food and Drug Administration. FDA approves first drug to treat hallucinations and delusions associated with Parkinson’s disease. United States Food and Drug Administration website. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm498442.htm. Published April 29, 2016. Updated May 2, 2016. Accessed July 8, 2016.
- Smith Y, Bevan MD, Shink E, Bolam JP. Microcircuitry of the direct and indirect pathways of the basal ganglia. Neuroscience. 1998:86:353–387.
- Meltzer HY, Roth BL. Lorcaserin and pimavanserin: emerging selectivity of serotonin receptor subtype-targeted drugs. J Clin Invest. 2013;123(12):4986–4991.
- Ballanger B, Strafella AP, van Eimeren T, et al. Serotonin 2A receptors and visual hallucinations in Parkinson disease. Arch Neurol. 2010;67(4):416–421.
- Munhoz RP, Moro A, Silveira-moriyama L, Teive HA. Non-motor signs in Parkinson’s disease: a review. Arq Neuropsiquiatr. 2015;73(5): 454–462.
- Center for Drug Evaluation and Research. NDA 207318 Pharmacology Review. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2016/207318Orig1s000Approv.pdf. Accessed October 20, 2016.
- Vanover KE, Robbins-weilert D, Wilbraham DG, et al. The effects of food on the pharmacokinetics of a formulated ACP-103 tablet in healthy volunteers. J Clin Pharmacol. 2007;47(7):915–919.
- Hunter NS, Anderson KC, Cox A. Pimavanserin. Drugs Today. 2015;51(11):645–652.
- United States Food and Drug Administration. ACADIA briefing information for the March 29, 2016 Meeting of the Psychopharmalogic Drugs Advisory Committee. United States Food and Drug Administration website. www. fda.gov. Published March 25, 2016. Approved March 29, 2016. Accessed July 8, 2016.
- Meltzer HY, Mills R, Revell S, et al. Pimavanserin, a serotonin (2A) receptor inverse agonist, for the treatment of parkinson’s disease psychosis. Neuropsychopharmacology. 2010;35(4): 881–892.
- Cummings J, Isaacson S, Mills R, et al. Pimavanserin for patients with Parkinson’s disease psychosis: a randomized, placebo-controlled phase 3 trial. Lancet. 2013;383:533–540.
- United States Food and Drug Administration. FDA briefing information for the March 29, 2016 Meeting of the Psychopharmalogic Drugs Advisory Committee. United States Food and Drug Administration website. www.fda.gov. Published March 2, 2016. Approved March 29, 2016. Accessed July 8, 2016.
- Acadia Pharmaceuticals. Efficacy and tolerability of Nuplazid™ (pimavanserin) in PD psychosis: analysis of integrated phase 3 placebo-controlled dataset [abstract]. Acadia® Pharmaceuticals. 2015. http://ir.acadia-pharm.com/phoenix.zhtml?c=125180&p=irol-newsArticle_Print&ID=2059705. Accessed July 8, 2016.
- Acadia Pharmaceuticals. Long-term effectiveness of Nuplazid™ (pimavanserin in PD Psychosis: data from 2 open-label studies [abstract]. Acadia® Pharmaceuticals. 2015. http://ir.acadia-pharm.com/phoenix.zhtml?c=125180&p=irol-newsArticle_Print&ID=2059705. Accessed July 8, 2016.
- Center for Drug Evaluation and Research. NDA 207318 Medical Review. United States Food and Drug Administration website. http://www.accessdata.fda.gov/drugsatfda_docs/nda/2016/207318Orig1s000MedR.pdf. Accessed October 20, 2016.
- Voss T, Bahr D, Jeffrey Cummings C, et al. Performance of a shortened Scale for Assessment of Positive Symptoms for Parkinson’s disease psychosis. Parkinsonism Relat Disord. 2013;19:295–299.
- Meltzer HY, Elkis H, Vanover K, et al. Pimavanserin, a selective serotonin (5-HT)2A-inverse agonist, enhances the efficacy and safety of risperidone, 2mg/day, but does not enhance efficacy of haloperidol, 2mg/day: comparison with reference dose risperidone, 6mg/day. Schizophr Res. 2012;141(2–3): 144–52.
- Friedman JH. Pimavanserin for the treatment of Parkinson’s disease psychosis. Expert Opin Pharmacother. 2013;14(14):1969–75.
- Pollack P, Tison F, Rascol O, et al. Clozapine in drug induced psychosis in Parkinson’s disease: a randomized, placebo controlled study with open follow up. J Neurol Neurosurg Psychiatry. 2004; 75:689–95.
- Factor SA, Feustel PJ, Friedman JH, et al. Longitudinal outcome of Parkinson’s disease patients with psychosis. Neurology. 2003;60(11):1756–1761.
- Pollak P, Destee A, Tison F, et al. Clozapine in drug-induced psychosis in Parkinson’s disease. Lancet. 1999;353: 2041–2042.
- Ondo WG, Tintner R, Voung KD, et al. Double-blind, placebo-controlled, unforced titration parallel trial of quetiapine for dopaminergic-induced hallucinations in Parkinson’s disease. Mov Disord. 2005;20(8):958–963.
- Rabey JM, Prokhorov T, Miniovitz A, et al. Effect of quetiapine in psychotic Parkinson’s disease patients: a double-blind labeled study of 3 months’ duration. Mov Disord. 2007;22(3):313–318.
- Shotbolt P, Samuel M, Fox C, David AS. A randomized controlled trial of quetiapine for psychosis in Parkinson’s disease. Neuropsychiatr Dis Treat. 2009;5:327–332.
- Clozapine REMS. Clozapine REMS Program website. https://www.clozapinerems.com. Accessed October 20, 2016.
- Love RC, Kelly DL, Feudenreich O, et al. Clozapine underutilization: addressing the barriers. National Association of State Mental Health Program Directors webite. September 2016. https://www.nasmhpd.org/sites/default/files/Assessment%201_Clozapine%20Underutilization.pdf. Accessed January 19, 2018.
- Merims D, Balas M, Peretz C, et al. Rater-blinded, prospective comparison: quetiapine versus clozapine for Parkinson’s disease psychosis. Clin Neuropharmacol. 2006;29(6):331–337.
- Friedman J, Lannon M, Comella C, et al. Low-dose clozapine for the treatment of drug-induced psychosis in Parkinson’s disease. N Engl J Med. 1999; 340:757–763
- Lexi-Drugs. Lexicomp. Wolters Kluwer Helaht, Inc. website. Riverwoods, IL. http://online.lexi.com. Accessed September 29, 2016.
- Redbook online. Thompson Readers. www.redbook.com. Accessed September 29, 2016.
- Clozaril (clozapine) [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2015.
- Seroquel (quetiapine) [package insert]. Wilmington, DE: AstraZeneca Pharmaceuticals LP; 2013.




{kind=link}
{kind=link}
{kind=link}
{kind=link}