

by Gahan Pandina, PhD; Joan Busner, PhD; Joseph P. Horrigan, MD; Christine McSherry, RN, BSN; Alison Bateman-House, MA, MPH, PhD; Luca Pani, MD; and Judith Kando, PharmD, BCPP
Dr. Pandina is with Janssen Pharmaceuticals in Titusville, New Jersey. Dr. Busner is with Signant Health in Blue Bell, Pennsylvania and Department of Psychiatry, Virginia Commonwealth University School of Medicine in Richmond, Virginia. Dr. Horrigan is Chief Medical Officer, AMO Pharma, Ltd. in Wonersh, United Kingdom and Consulting Associate Professor, Duke University in Durham, North Carolina. Ms. McSherry is Founder, Jett Foundation, and Co-founder and Chief Executive Officer, Casimir, LLC in Plymouth, Massachusetts. Dr. Bateman-House is with New York University Grossman School of Medicine in New York City, New York. Dr. Pani is former Director General, Italian Medicines Agency (AIFA), former Committee for Medicinal Products for Human Use (CHMP) member, and former Scientific Advice Working Party (SAWP) member, European Medicines Agency (EMA); he is with the University of Miami in Miami, Florida, and VeraSci in Durham, North Carolina. Dr. Kando is with Tris Pharmaceuticals in Monmouth Junction, New Jersey.
Funding: No funding was provided for this article.
Disclosures: Dr. Pandina is a full-time employee of Janssen Research & Development, LLC, and a Johnson & Johnson stockholder. Dr. Busner is a full-time employee of Signant Health and may own stock/stock equity. Dr. Horrigan is an employee of AMO Pharma, Ltd. Ms. McSherry holds multiple consultancies and collaborations with industry and academia. Dr. Bateman-House is a Voting Member, COVID-19 Data Safety Monitoring Board, New York University Langone Health, and an Ethics Board Member of Alexion Pharmaceuticals. Dr. Pani is a part-time employee of the Universities of Modena and Reggio Emilia in Italy and Miami, FL, US, and acts as Chief Scientific Officer for the EDRA-LSWR Publishing Company and the Inpeco SA Total Lab Automation Company; he is also Vice President for Regulatory Strategy and Market Access Innovation at VeraSci in Durham, NC, US. He has previously acted as a scientific consultant for AbbVie USA; Acadia USA; BCG Switzerland; Boehringer Ingelheim International GmbH; Compass Pathways, UK; EDRA-LSWR Publishing, Italy; Ferrer Spain; Inpeco SA Total Lab Automation Company, Switzerland; Johnson & Johnson USA; NeuroCog Trials USA; Otsuka USA; Pfizer Global USA; PharmaMar Spain; Relmada USA; Takeda, US; VeraSci, US; and Vifor, Switzerland. Dr. Kando is an employee of Tris Pharmaceuticals and has stock in Johnson & Johnson, Takeda, and McKesson.
Innov Clin Neurosci. 2023;20(1–3):18–24.
Abstract
This article expands upon a session, titled “Implications of Pediatric Initiatives on CNS Drug Development for All Ages—2020 and Beyond,” that was presented as part of a two-day meeting on pediatric drug development at the International Society for Central Nervous System (CNS) Clinical Trials and Methodology (ISCTM) Autumn Conference in Boston, Massachusetts, in October 2020. Speakers from various areas of pediatric drug development addressed a variety of implications of including children in drug development programs. The speakers wrote summaries of their talks, which are included here. The session’s lead chair was Dr. Gahan Pandina, who wrote introductory and closing comments. Dr. Joseph Horrigan addressed the current landscape of pediatric development programs. Dr. Gahan Pandina addressed how the approach to research in pediatric populations affects the drug development process and vice versa. Dr. Alison Bateman-House discussed the ethical implications of research in the pediatric population. Dr. Luca Pani discussed some of the global regulatory issues and challenges concerning research in pediatric patients. Dr. Judith Kando served as a discussant and posed new questions about means of facilitating pediatric research. Finally, Dr. Gahan Pandina provided closing comments and tied together the presented issues. This paper should serve as an expert-informed reference to those interested and involved in CNS drug development programs that are aimed at children and/or required, through regulations, to include children as part of the approval process.
Keywords: Pediatric drug development, CNS pediatric psychiatric drug development, pediatric neurologic drug development, pediatric clinical trials, regulatory aspects of pediatric drug development
Introductory Comments—Gahan Pandina, PhD
Pediatric research is critical to addressing the significant unmet medical needs manifesting in orphan disease populations. Early strategic planning in drug development typically includes pediatric populations, but pediatric studies are not started until after the completion of Phase I studies in adults. This series of talks addressed some of the challenges inherent in various aspects of pediatric drug development and what considerations should be accounted for to enable earlier, high-quality pediatric research. Topics addressed in this series range from strategic planning for pediatric research and outcome measures to Institutional Review Board (IRB)/ethics committee requirements and regulatory considerations.
Introduction to Pediatric Aspects of Drug Development: The Current Landscape—Joseph P. Horrigan, MD
There is a general understanding that many, if not most, neurological and psychiatric disorders can begin during childhood and/or adolescence or can have variations that present early in life. The pediatric presentations of these disorders are often understudied. When they occur early in life, these disorders can cause lifelong morbidity, especially if diagnosis is delayed or if the disorder is treated insufficiently. For example, anxiety disorders are ubiquitous but also notorious for a delay in diagnosis and treatment. Consequently, untreated anxiety disorders may become almost intractable and can significantly alter the life course of youth as they move into adulthood, despite the fact that anxiety disorders often respond robustly to appropriately chosen medications, as well as psychotherapies, such as cognitive behavioral therapy (CBT).
Many central nervous system (CNS) disorders are both familial and subject to genetic anticipation, affecting multiple generational levels of a family with progressively increasing severity. In practice, this can mean that the clinician may be dealing with affected parents when formulating and implementing a treatment plan for the parents’ affected children and adolescents.
Similarly, clinicians that specialize in the treatment of adults may quickly uncover pediatric manifestations of the disorder when they conduct a review of the pedigree of the patient. Type 1 myotonic dystrophy and bipolar disorder are two premier examples in this regard. Early intervention and treatment of CNS disorders holds the prospect of altering the disorder’s trajectory and, in doing so, assuring better outcomes.
In terms of pharmacotherapies, virtually all approved CNS medicines are eventually prescribed to children and/or adolescents, especially when the youth’s clinical presentation is complex or has culminated in an evaluation in a tertiary care setting. Recent pediatric regulatory initiatives are playing a role in ameliorating this situation by both requiring and incentivizing clinical trials that may more effectively inform prescribing practices in younger affected individuals.
Pediatric clinical studies, like adult studies, evaluate safety and effectiveness, although clinical trials involving children and adolescents are often mindful to assess, either directly or indirectly, organs and organ systems that are undergoing rapid developmental change, including bones, the brain, and reproductive organs. In practice, pediatric clinical trials often include assessments, such as dual-energy x-ray absorptiometry (DEXA) scanning to assess bone growth, and brief bedside cognitive assessments, such as the National Institutes of Health (NIH) Toolbox.
In addition, efficacy assessments in these studies may rely more heavily on observable behaviors or features of the disorder under study and/or on direct caregiver reports due to the limited insight and expressive language abilities of younger patients with neurological and psychiatric disorders. Also, the phenotypic presentations in affected youth may be more directly amenable to observation. Examples include attention deficit hyperactivity disorder (ADHD), in which youth are often more overtly hyperactive than adults, and affective disorders that, when severe, can be accompanied by flagrant psychotic phenomena that may be readily apparent in affected youth.
An important disadvantage of relying on parent or caregiver reports is the proclivity towards expectancy, which can lead to inflated placebo response rates, especially in clinical trials involving younger children, in which parental ratings are particularly vulnerable to the influence of their own hopefulness and gratitude, and this needs to be managed proactively. To counter this response, it is very important to engage parents and caregivers as research partners and encourage them to stand shoulder-to-shoulder with clinician-raters in assessing treatment-related changes with a disciplined eye.
Despite these challenges, pediatric drug development is an important and worthwhile endeavor. When pediatric clinical trials are designed and conducted in a thoughtful way, they can culminate in key insights occurring earlier in drug development, and they may offer an opportunity to develop a treatment that improves functioning and quality of life much earlier in the patient’s lifespan than might otherwise have been considered possible.
Drug Development: The Chicken or the Egg? How the Approach to Research in Pediatric Populations Affects the Drug Development Process and Vice Versa—Gahan Pandina, PhD
Most CNS disorders begin, at least in a subpopulation, in childhood. Despite this fact, concerns about conducting clinical trials in pediatric populations have resulted in a limited amount of pediatric research. Some of these concerns may be valid, including potential safety/tolerability issues, complexity of performing medical or clinical procedures in children, a lack of well-designed and validated pediatric endpoints, and difficulties with the consent process. However, there are other common, but often unconsidered, reasons for the lack of pediatric studies, such as the common drug development strategy of performing juvenile toxicology studies late in the development cycle due to the potential associated complexity and costs.
Pediatric legislation in the United States (US) and European Union (EU) has both required and incentivized pediatric drug research. Food and Drug Administration (FDA) legislation, such as the Pediatric Rule (1998), the Best Pharmaceuticals for Children Act (BPCA; 2002) and the Pediatric Research Equity Act (PREA; 2003), have resulted in increased numbers of studies among pediatric patients. Data from the FDA tracking database, Pediatric Labeling Changes,1 show labeling changes associated with pediatric studies conducted as part of this legislation (Figure 1). While pediatric labeling may lag behind the initial approval for drugs, published literature confirms an increase in pediatric trials, and subsequent approvals and labeling have increased substantially over the past 20 years.2
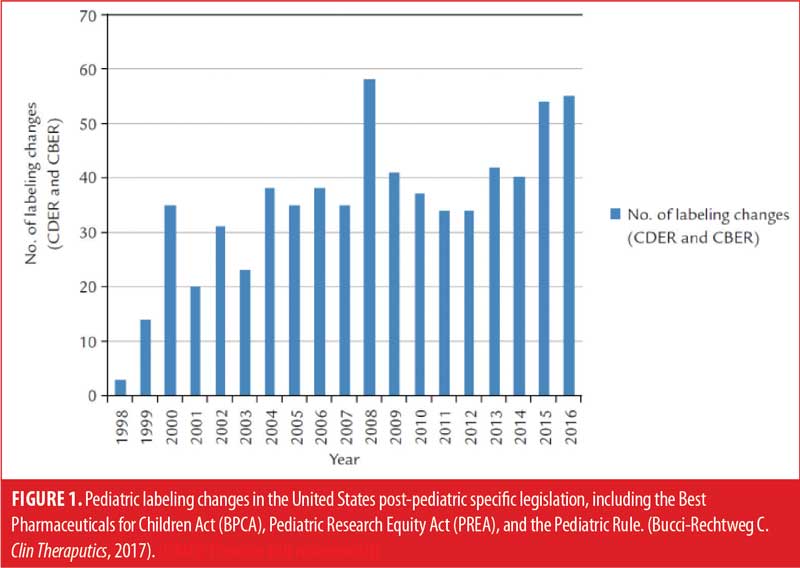
Several issues that affect the timing and conduct of pediatric studies must be considered. Across most therapeutic areas, and particularly among psychiatry and neurology trials, unless the condition or disorder under study is primarily a pediatric condition, early safety studies are typically conducted in adult populations. This is due to many factors, including the lack of global consensus on many diagnoses in childhood, limited availability of pediatric research centers, problems with outcome measures or standard assessment procedures in the pediatric population, more complex research procedures (e.g., consent/assent, risk tolerance, protocol complexity, etc.), and more difficult study conduct. Despite these factors, the pediatric population may be more amenable to therapeutic intervention, having greater symptomatic benefit and functional outcome.
One complexity in pediatric research is the presence of discrepancies in legislation about when pediatric data are required and when the research plan including pediatric data must be submitted for consideration by the FDA or European Medicines Agency (EMA). For the EMA, this date is not later than at the completion of the human pharmacokinetic (PK) studies, while for FDA, it is not later than 60 days after the end-of-Phase II meeting. In addition, pediatric populations may require independent PK studies and/or formulations, which can add to development timelines and costs. While this can be managed, it must be considered very early in development—earlier than typically considered in the standard timeline—in order to be properly prepared. The requirement for juvenile toxicology studies prior to a pediatric study also must be planned in early development, as these data are considered relevant for safety in human children.3
In order to facilitate and encourage earlier pediatric drug development, a number of strategies can be employed, which include the following:
- Establish and support quality pediatric networks.
- Highlight the importance and clinical value of pediatric indications.
- Obtain global alignment on endpoints, including for novel diseases/disorders.
- Engage regulators early to discuss research approaches.
- Discuss timing and approaches to obtain juvenile toxicity data.
- Use extrapolation techniques with existing evidence (modeling and simulation).
- Use short-term, biomarker driven studies to establish proof-of-concept (PoC) efficacy.
Facilitation of pediatric research early in the drug development process can have a major impact on human health, including improved morbidity and mortality. Increasing focus and investment in pediatric drug development requires collaboration among diverse stakeholder groups. The research community, private industry, and regulators can continue to collaborate in public forums to formulate better approaches to pediatric drug development.4
Unique Issues in Outcome Measurement When Including Pediatric Populations—Christine McSherry, RN, BSN
There are factors within pediatric clinical trials that warrant unique considerations and discussions. In particular, there are concerns about the efficacy of current outcome measurement tools used for pediatric clinical trials due to the lack of sensitivity to change, lack of specificity, overgeneralization of use, and lack of insight on and incorporation of affected communities, leading to questions about the saliency of measures and variables in measurement tools.
Many factors are considered when designing a pharmaceutical or other medical intervention efficacy study; such factors warrant more consideration for those studies involving a pediatric, or other especially vulnerable, population. First and foremost, the ideal impetus for developing and creating clinical interventions is to improve the lives of those affected by diseases, disorders, and other ailments or illnesses. These diseases, disorders, and other ailments and illnesses often affect more than just an individual, especially when a pediatric population is affected. As such, direct partnerships with patients, parents, caregivers, and disease communities become integral.
Patients and other participants are more than subjects; they are repositories of data and the vehicle through which data transforms into meaning. Without centrally orienting the participant and other affected individuals in trials and studies, there is a loss in the context for determining appropriate outcomes and efficacy. Direct partnerships facilitate participants as more than subjects; they allow direct input from participants on potential improvements in symptoms and on what improvements are meaningful, both subjectively and clinically. Additionally, structural support, mainly via critical paths of guidance from regulatory agencies, is inherently necessary. These pathways are needed to develop novel methods, measurement tools, and interventions to support clinical studies. The availability of reliable, valid, and disease-specific outcome measures and tools is another constitutive factor for studies. There is a strong need for novel methods and measurement tools due to the rapid development of new interventional therapies for rare diseases and other diseases that have lacked any interventional avenues until the present decade. In the past few years, there have been two dozen clinical drug trials involving pediatric populations affected by Duchenne muscular dystrophy (DMD) that failed to conclude with any definitive answers. Clinical outcome measures, surveys, and patient-reported outcomes (PROs) designed for a broad swath of disorders and diseases often lack sensitivity to change and disease specificity and overlook which measures or variables are essential to participants.
Casimir was born out of concerns about these limitations in pediatric outcome measurement tools, a problem precipitated from the direct experience of our founding members. As part of the rare disease community, our founding members became familiar with the aspects and process of participation in clinical trials: traveling, scheduling, and financial requirements, as well as the hopes and disappointments. Our personal clinical trial experiences exposed gaps in the way treatments for rare diseases are investigated and approved by regulatory agencies and the lack of patient and participant input into these trials and review processes. Our experiences in DMD trials led to conversations with stakeholders, clinicians, and regulatory agents. Insight from these conversations led us to reach out to Dr. Janet Woodcock, Director of the FDA’s Center for Drug Evaluation Research (CDER), and other CDER officials in the Division of Neurology to discuss our concerns in July 2013. Our initial address and discussion with the FDA and CDER about incorporating patient perspectives and experiences into clinical trials was met with an open reception. From these discussions, Dr. Woodcock and CDER requested video and other supportive evidence regarding patient experiences. By June 2013, we had gathered a set of video clips expressing participants’ experiences in the trial for Sarepta’s drug, eteplirsen. Pleased with our data, CDER requested that we continue our efforts and gather additional videos and interviews with participants to ascertain what outcomes are important to them and how to meld their experiences and perspectives with activities of daily living (ADLs) and functional abilities, and to do so in a manner that is both measurable and quantifiable.
Beginning our efforts in the last quarter of 2014, we systematically gathered data from young participants in the efficacy trial for eteplirsen, conducting semi-structured interviews to gain insight into their experiences and learn what ADLs were important to them. From this participant insight, we then gathered video clips of participants performing these identified ADLs. In July 2015, we compiled a report on our data, analysis, and findings and presented it to CDER, with a final report given to the FDA and Sarepta in April 2015. The results of this report were used as part of the FDA’s review for a new drug application (NDA) for eteplirsen.
From this experience and discussion with the FDA, Casimir established an aim to develop meaningful outcome measures, with a focus on learning from the participant. Using a mixed-methods approach, combining qualitative data via interviews and quantitative data via video capture of functional movements, we aim to contextualize outcome measures and tailor them to specific disease communities. Using mixed methods allows for the incorporation of patient and caregiver experiences and perceptions of change directly into the development of outcome measures, taking into account what they feel is functionally important and what may be a meaningful change. This more comprehensive approach aims to provide sufficient rigor to incorporate qualitative data toward the totality of data in determining efficacy in clinical trials. Furthermore, we aim to develop tools that would allow the clinical trial experience into the lives of patients and families. We also aim to create a centralized, home-based outcome measures tool that would capture functional ability in a comfortable home setting and mitigate any potential confounding variables due to travel fatigue, stress from unfamiliar environments, and variations in clinical instructions, as well as lessening the time and financial burden on participants and their families and opening up access to clinical trials to a larger population.
Overall, we advocate for stronger partnerships and more commingled relationships among multiple entities and stakeholders in clinical trials. The relationship between pharmaceutical companies, researchers, structural and regulatory agents, patients, caregivers, and other affected disease community members must be closely aligned to benefit patients, affected communities, and other stakeholders.
IRB/Ethics Committee Requirements for Pediatrics and How These May Affect Drug Research—Alison Bateman-House, MA, MPH, PhD
Most countries impose protections on pediatric research beyond those required for research on adults, who are deemed able to make informed and noncoerced decisions for themselves. These added protections may incline new drug sponsors to avoid or delay pediatric studies of their products. While additional ethical, regulatory, and compliance requirements were intended to protect children from exploitation, an unintended consequence has been the comparative neglect of pediatric research, resulting in a dearth of evidence-based treatments for children. This, is turn, leads to disadvantages for the health and wellbeing of children who, for example, may be treated with interventions that were only tested on adults and for which pediatric dosage or use guidelines may be based on extrapolation or educated guesses, rather than data.
Ethical standards call for research on humans to be transparent, conducted on informed volunteers (in almost all circumstances), and designed to yield maximum benefit and minimum risk. While the risks most often fall on those who participate in the research, the benefits can be experienced either by the research volunteers or society at large, often via the advancement of knowledge on a subject. Ethics bodies on the institutional, regional, or national level may be charged with reviewing research on humans and creating policies that might be either voluntary or compulsory. Alternatively, requirements for research on human subjects may be codified into laws and regulations. Researchers therefore must be aware of relevant, legally mandated policies and ethical guidance or discussions that may not have the force of law. Both of these—legally enforceable rules and those that do not have legal weight—tend to vary between nations, funders, and institutions, among others. As such, not only do researchers need to be aware of a complex array of policies, they must also be aware that these policies may well differ depending on the situation, something that may cause inconveniences and serious obstacles. As an example, there is no international consensus on what constitutes pediatric research; nations vary in how narrowly they construe this category. Thus, conducting a clinical trial internationally may result in differing age eligibilities at various trial sites, in accordance with local laws.
Where there are no firm rules, there are open questions to be navigated. For example, while ethics may require that research yield maximum benefit, who gets to decide what counts as a benefit? When a research activity poses both anticipated benefits and risks, who gets to evaluate the risk/benefit ratio and decide if it is acceptable? If ethics says that individuals should be given the opportunity to make their own voluntary and informed research decisions, what does that mean for research on children, who are not legally able to consent to research? Some of these questions have straightforward answers; for example, there is global consensus that specified adult decision-makers have the legal standing to grant permission to conduct research on their child. Other, less settled, questions—such as how much benefit and risk are acceptable when seeking to test a new drug in children—entail the involvement of, at minimum, ethics, legal, and regulatory representatives, and often also entail consultation with the relevant patient community or advocates who speak on that community’s behalf.
In order to ensure that children are not deprived of the benefits that stem from research, there is an ethical obligation to conduct pediatric research, even when this requires additional effort on the part of researchers. Reaching out proactively to ethics bodies, regulators, and patient communities is a worthwhile way for research sponsors to design studies that will be both maximally acceptable to potential participants (and their caregivers) and designed to evaluate interventions in terms of the clinical endpoints deemed most important by not only regulators, but also patients and payors.
Regulatory Challenges When Including Pediatric Populations: A Comment on the European Perspective—Luca Pani, MD
Background. When it comes to pediatric regulation in drug development, the EMA and FDA have more similarities than in other areas of interest and competence, with notable differences that descend from their respective reference legislations. This section addresses the EU perspective.
The major objective of the EU Pediatric Regulation is to improve the health of children by:
- Increasing high quality, ethical research into medicines for children
- Increasing availability of authorized medicines for children
- Increasing information on medicines.
To achieve the above objective, the EMA demands that no unnecessary studies are performed on children and that a pediatric indication for any given pharmaceutical product is evaluated without delaying authorization for the same product in adults.
Applicants should be reminded that in Europe, pediatric development (i.e., the Pediatric Investigation Plan [PIP]) is mandatory for any new product, unless product-specific or class waivers/deferrals for specific conditions and dosage forms are granted. To respect the provision that adult authorization should not be postponed, studies in children can be deferred, initiated, and/or completed after applying for marketing authorization in adults.
For existing products, the PIP becomes mandatory if the Marketing Authorization Holder applies for a new indication, route, or dosage form, and if the product is protected by a patent/supplementary protection certificate (SPC). In this case, waivers and deferrals are also possible, as are incentives for voluntary development (e.g., pediatric-use marketing authorization [PUMA]) and additional obligations to report studies (art. 46 of the EU Pediatric legislation). Table 1 compares the US PREA and BPCA with the EU PIP.
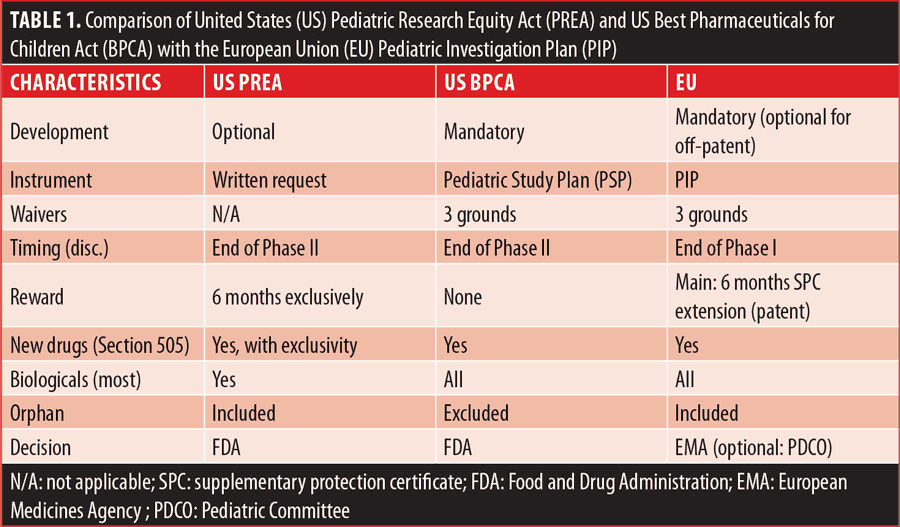
A complex system of incentives and obligations regulates the pediatric EU drug development. For instance, a reward is always given for all EU PIPs correctly completed, but PIPs are always required, unlike the US PREA, where there is an obligation without reward. Incentives are provided in the EU if the pediatric development is compliant with agreed PIP (compliance statement in the marketing authorization), if results of studies are included in Summary of PC + patient’s leaflet, or if product is authorized in all member States.
Incentives could also be foreseen for nonorphan products with a six-month extension of the SPC (patent protection) or orphan medicinal products with over two additional years of market exclusivity and eight and two years of data and market protection, respectively, with PUMA. It is worth mentioning that a product-specific or class waiver or inconclusive studies in PIP do not trigger the reward, while negative results of a well-completed PIP may allow a reward.
Pediatric Committee (PDCO). The PDCO is the committee responsible for activities on medicines for children and supporting the development of such medicines in the EU by providing scientific expertise and defining therapeutic needs in pediatrics. The PDCO was established in line with the Pediatric Regulation (EC) No 1901/200611, which came into force in 2007.
The PDCO’s main role is to assess the content of PIPs, which determine the studies that companies must conduct in children when developing a medicine. More precisely, the PDCO adopts opinions on PIPs/waivers (these decisions are signed directly by the EMA Executive Director, not by the European Commission) and provides advice on any question relating to pediatric medicines (advice is given at the request of the EMA’s Executive Director or the European Commission). In addition, an essential role of the PDCO is to assess data generated in accordance with agreed PIP and adopt opinions on the use of medicines in the EU pediatric population (advice is given at the request of the Committee for Medicinal Products for Human Use [CHMP] or any national competent authority).
In general, the PDCO adopts detailed opinions on the quality, safety, or efficacy of a medicine for use in the pediatric population and advises the CHMP, Member States, and the EMA Executive Director on the content and format of data to be collected through investigations into the use of medicines in children. However, the PDCO does not address the marketing authorization applications, which are the direct responsibility of the CHMP.
EU PIP. A PIP is deemed necessary every time that data on efficacy, safety, and age-appropriate formulations are needed. Timelines for the start and completion of each study should be illustrated, and a detailed description and analysis on each condition/indication and formulation, as well as for each pediatric population subset (not only age groups), must be discussed. A PIP application should cover the following topics: formulation, toxicology, PK, pharmacodynamics (PD), carcino, genotox, juvenile animals, safety, PoC, dose-finding, and any efficacy or safety issues of potential interest.
A PIP should be requested as early as at the end of Phase I, while amendments to it can be proposed and discussed with the PDCO until the end of Phase III, when a compliance check will be run before marketing authorization can be granted. As previously noted, a deferral in this timeline could be requested, providing that it is justified. There are several conditions where a PIP is not required. Off-patent products already authorized in the EU and authorized products that do not have a valid SPC or a valid patent that qualifies for it do not need a PIP. New medicinal products that belong to certain, specific groups, such as traditional herbal medicinal products, homeopathic products, generic products, hybrid products, and biosimilar products do not need a PIP. Finally, when there is a waiver either for a class of products in a condition or for all products in a condition, a PIP is not requested. The EU Legislation comprises three types of waivers: 1) Total (product-specific) waiver for all pediatric subsets (in one or more specific conditions); 2) partial waiver for one or more subset(s) or indication(s), but not for others where the PIP is still required and 3) class waiver for a class of medicinal products in a condition.
The legal grounds that support a PIP waiver are lack of efficacy and safety in the pediatric population, the indication for a disease or condition occurring only in adults, and the lack of significant therapeutic benefit.
The deferral is an EU regulatory instrument that is employed to avoid delaying marketing authorization in adults. Being deferred means that the marketing authorization application for adults is possible before initiation/completion of one or more studies/measures in the PIP. The deferrals can be given on specific study/measure (e.g., US PREA: “total” deferral) or for initiation and/or completion of the study/measure. The completion of a clinical trial may be deferred, whereas the initiation may not be if a waiver has not been granted, and the completion dates must be established in any case of deferral.
Additional considerations. In August 2020, the European Commission published its assessment of legislation for medicines for rare diseases and children. This legislation, therefore, should also be read in conjunction with that dedicated to rare diseases in Europe. The assessment of the two regulations—the pediatric and orphan drugs regulations—is the first to be carried out since they were adopted. The reason they have been assessed together is because most rare diseases can occur in children and many diseases in children are rare. The highlights of the evaluation can be found on the European Commission’s website, to which reference should be made.5
The review was prompted by the findings of independent studies, reports from the European Commission and EMA, and information from public stakeholder consultations. The evaluation found that both regulations have facilitated the development and availability of medicines for patients with rare diseases and children, allowing private and public investments to be directed toward research in these areas.
The number of authorized molecules has increased, and the off-label use of medicines approved for adults and prescribed for children has decreased. However, the evaluation also found that both regulations failed to adequately support the development of specific products in areas of greatest unmet medical need, confirming that pharmaceutical companies, in general, tend to develop products in the most profitable therapeutic areas.
In the EU Pediatric Regulation, therefore, systems should be overseen that allow clinical trials to be directed toward areas of greater medical need for children, whereas, to date, studies have been driven by medical needs for adults, and the cost of conducting clinical trials in children could instead be compensated for by extending the duration of the patent (SPC), see Article 29 of the pediatrics EU legislation.
This type of procedure can be initiated by the holder of a marketing authorization when requesting a new indication, a new pharmaceutical form, or a new route of administration for the use in children of a product authorized under Directive 2001/83/EC.
With respect to the Orphan Drug Regulation, the evaluation found that incentives remain relevant to encourage drug development, but many of these are increasingly similar to standard drugs, and therefore, 10-year market exclusivity could be questionable in such cases. The Orphan Drug Regulation allows for a reduction of the period of market exclusivity once a medicine has been commercially successful, but in practice, EU Member States have not activated the procedure because it is too difficult to support such a decision with rigorous evidence.
In addition, the European Commission’s assessment highlighted that those medicines developed through the two regulations are not accessible to all patients in the same way in all Member States. This is primarily due to external factors, such as how the regulations are implemented, strategic launch decisions by pharmaceutical companies, and the influence of national policies on reimbursement systems and pricing, and it will most likely be addressed in the foreseeable future.
An essential reference is included at the end of this article.5
Expanded Discussion on Other Needs to Facilitate Peds-first Drug Development Approach—Facilitator/Discussant, Judith Kando, PharmD, BCPP
When conducting research in adults, wherein findings can be readily applied to the day-to-day practice of managing patients, one faces innumerable challenges. These challenges can be even more difficult to overcome when the population under evaluation involves children. As a result, the quality and number of pediatric drug research trials has lagged behind trials involving adults.6 When pediatric trials are conducted, it often occurs after trials in adults have been completed. We are asking researchers to consider a pediatric-first drug development approach.
There have been legislative and regulatory approaches that attempted to address pediatric drug development, including improving the licensing and labeling of products. Examples of these are the BCPA and PREA. The BCPA became law in 2002. It was reauthorized in 2007 and became permanent in 2012, with some modifications, in Title V of the FDA Safety and Innovation Act (FDASIA). The goals of the BCPA are to encourage the pharmaceutical industry to perform pediatric studies to improve labeling for patented drug products used in children by granting an additional six months of patent exclusivity. In addition, the NIH can prioritize therapeutic areas and sponsor research, including clinical trials for off-patent drug products that are believed to hold promise in the treatment of children.7 The PREA gives the FDA the authority to require pharmaceutical companies to conduct pediatric studies as part of the postapproval commitment. Appropriate formulations for each age group must be evaluated.8 Like the BCPA, this act was also made permanent in 2012, with modifications.
Since the legislation was passed, significant progress has been made. In a 2016 report to Congress, then FDA Commissioner, Robert Califf reported that “the successful completion of pediatric studies under BPCA and PREA has led to the addition of new pediatric information on labeling for over 600 products since the enactment of these two laws.”9 There have been additional impacts, including the generation of science around the differences between children and adults and understanding how these differences impact drug and biological product development. Progress has also been made in understanding pediatric study endpoints, biomarkers, and exposure-response relationships.9 Despite our progress, studies in neonates, infants, childhood rare diseases, and disorders that are routinely diagnosed in childhood remain a challenge.
Many areas of opportunity exist. For instance, pediatric trials often occur after adult trials have completed. If we were to consider the pediatric trials at the same time as we are designing the adult trials, specific considerations could be taken into account, such as unique dosage formulations, safety and toxicity concerns, and high or unusual response in those receiving placebo. Moreover, there are disorders that occur primarily in childhood or occur early in childhood that require drug discovery and development programs aimed specifically at these disorders. In order to conduct valid and meaningful research in the child population, there is a need for researchers with a unique and complex set of skills. These individuals require specific training to be able to identify subtle but meaningful clinical changes. Importantly, we need to find a way to conduct research in this population that involves a geographically diverse population, not just subjects who are geographically close to trial sites or who are willing/able to consistently travel long distances.
In summary, significant progress has been made in advancing research in children, which has generated data that guide clinicians in the treatment of childhood disorders and guide the direction of future research. There are many areas of opportunity to continue to progress and build upon the advancements that have been made following the enactment of the pediatric specific legislation, including the BCPA and PREA.
Closing comments—Gahan Pandina, PhD
Pediatric research requires technical skills and experience, as well as specialized approaches, to enable better research outcomes for orphan diseases. Strategic approaches to pediatric research should be part of early program planning to maximize efficiency and gather pediatric data as early as possible. Engagement with stakeholder groups, including patients, caregivers, advocacy groups, clinicians, and multidisciplinary researchers, is crucial. Ethics and IRBs require special processes and procedures to ensure that adequate protections are in place prior to research conduct. Special considerations should be given to the selection of outcome measures, including patient-centric approaches, to ensure that these measure clinically meaningful variables that are sensitive to potential drug effects. There are additional opportunities to engage with regulatory authorities around pediatric programs; these interactions can lessen risk and better inform trial designs for drug development in orphan diseases.
Authors’ Note
Drs. Pandina and Busner served as Co-chairs of the Pediatric Initiatives Session. Drs. Horrigan, Bateman-House, Pani, and Kando and Ms. McSherry served as invited speakers.
References
- United States Food and Drug Administration. Pediatric labeling changes. Current as of 30 Aug 2022. https://www.fda.gov/science-research/pediatrics/pediatric-labeling-changes. Accessed 18 Jan 2023.
- Hudgins JD, Bacho MA, Olsen KL, Bourgeois FT. Pediatric drug information available at the time of new drug approvals: a cross‐sectional analysis. Pharmacoepidemiol Drug Saf. 2018;27(2):161–167.
- Fisher JE Jr, Ravindran A, Elayan I. CDER experience with juvenile animal studies for CNS drugs. Int J Toxicol. 2019;38(2):88–95.
- Ollivier C, Mulugeta Y, Ruggieri L, et al. Paediatric extrapolation: a necessary paradigm shift. Br J Clin Pharm. 2019;85(4):675–679.
- European Commission. Evaluation of the medicines for rare diseases and children legislation. https://ec.europa.eu/health/human-use/paediatric-medicines/evaluation_en. Accessed 18 Jan 2023.
- Zylke JW, Rivara FP, Bauchner H. Challenges to excellence in child health research: call for papers. JAMA. 2012;308(10):1040–1041.
- Eunice Kennedy Shriver National Institute of Child Health and Human Development. Best Pharmaceuticals for Children Act (BPCA). https://www.nichd.nih.gov/research/supported/bpca. Accessed 18 Jan 2023.
- United States Food and Drig Administration. Pediatric Research Equity Act | PREA. Current as of 7 Nov 2019. https://www.fda.gov/drugs/development-resources/pediatric-research-equity-act-prea. Accessed 18 Jan 2023.
- Califf RM. Best Pharmaceuticals for Children Act and Pediatric Research Equity Act: July 2016 Status Report to Congress. Department of Health and Human Services, Food and Drug Administration. Jul 2016. https://www.fda.gov/media/99184/download. Accessed 18 Jan 2023.