
by Steven T. Szabo, MD, PhD; Bruce J. Kinon, MD; Stephen K. Brannan, MD; Andrew K. Krystal, MD, MSc; Joop M A van Gerven, MD, PhD; Atul Mahableshwarkar, MD; and Gary S. Sachs, MD
Dr. Szabo is with Duke University Medical Center, Durham, North Carolina, and Veterans Administration Medical Center, Durham, North Carolina; Dr. Kinon is with Lundbeck LLC, Deerfield, Illinois (Dr. Kinon was with Eli Lilly and Company, Indianapolis, Indiana, when this material was presented); Dr. Brannan is with Takeda Global Research & Development Center, Inc., Deerfield, Illinois; Dr. Krystal is with Duke University Medical Center, Durham, North Carolina; Dr. van Gerven is with Centre for Human Drug Research, Leiden, The Netherlands; Dr. Mahableshwarkar is with Takeda Global Research & Development Center, Inc., Deerfield, Illinois; Dr. Sachs is with Massachusetts General Hospital, Boston, Massachusetts.
Innov Clin Neurosci 2015;12(3–4 Suppl A):11S–25S.
Funding: No funding was provided for this article.
Financial disclosures: Dr. Szabo is a consultant for Otsuka Pharmaceutical. Dr. Kinon was an employee of Eli Lilly and Company when this material was presented. Dr. Brannan and Dr. Mahableshwarkar are employees of Takeda Pharmaceuticals. Dr. Krystal has received grant support from Astellas Pharma Inc., Janssen Pharmaceuticals, Inc., Novartis Corporation, Sunovion Pharmaceuticals Inc., and Teva Pharmaceuticals USA and serves as a consultant to AstraZeneca, Eisai Inc., Jazz Pharmaceuticals, Inc, Johnson and Johnson, Merck & Co, Roche, Somnus Therapeutics Inc, Teva Pharmaceuticals USA, and Vantia Ltd. Dr. Sachs owns stock, stock options, or bonds in Concordant Rater Systems, LLC; has received grant support from GlaxoSmithKline, AstraZeneca, Wyeth, Eli Lilly, Sanofi, and Abbott Laboratories; and has served as advisor or consultant for GlaxoSmithKline, Pfizer, AstraZeneca, Wyeth, Eli Lilly, Sanofi, and Abbott Laboratories.
Key words: Clinical trials, experimental design, methodology, genetic, research domain criteria, RDoC, proof-of-concept
Abstract: Once a molecule has been characterized as engaging an identified target at the appropriate location (affinity and potency), the next step involves designing experiments that will determine its pharmacodynamic activities both for efficacy (on target) and safety-tolerability (on/off target). Two expert presentations focused on looking back at completed programs and two concentrated on looking forward at ongoing programs. Specific discussions pertain to assessment of pharmacologic agonists (mGluR2/3, k-opiate, peroxisome proliferator-activated receptor gamma) and antagonists (orexin and cannabinoid) in disorders of cognition, mood, and anxiety. Advanced experimental study designs using genetics to guide a treatment trial in Alzheimer’s disease and neural target-based approaches as the primary outcome measure in the National Institute of Mental Health-sponsored Fast-Fail Trials (FAST)-Mood and Anxiety Spectrum Disorders (MAS) initiative for depression showcases novel methodological approaches. Of interest, some of these initiatives were successful, while others were not, and two are currently ongoing. In conclusion, methodologies that were utilized and are currently employed to reach a successful clinical drug trial outcome are appreciated, and in case of failure, approaches to reviewing programs to enable learning that would be helpful to future programs are brought forth. This article is based on proceedings from the “Designing the Right Series of Experiments” session, which was held during the International Society for Clinical Trials Meeting (ISCTM) in Philadelphia, Pennsylvania, September 30 to October 2, 2013.
Introduction
Methodological shortcomings have likely contributed to failed drug development in psychiatry, and intense scrutiny in feasibility of continuing these efforts is currently ongoing. The ability to design the right series of experiments is as important as formulating the correct question to be answered.
Controlling for as many variables as possible in experimental design is a mainstay often thought to lead to less variance and success. However, in clinical trials using therapeutics for complex central nervous system (CNS) disorders, a number of subjective variables are monitored and considerable variance in patient populations occur, in part due to diagnostic impression, which may lead to diluted treatment effects and diminished potential for delivery on their promised mechanism. Even prior to the stage of human clinical trials, preclinical investigations can offer great insights into the potential mechanism of action and efficacy of compounds in treatment of CNS disorders. However, given the complexity of neuropsychiatric illnesses with translational efforts having inherent imprecision their applicability is sometimes questioned. This article focuses on evaluating the scientific process once a compound meets the current rigors of preclinical evaluation and is ready to undergo testing in humans. Creating methodologically intact clinical trials to test a known medication target in humans, or even a proof of principle agent (i.e., hypotheses driven), carries forward important considerations that at times are specific to the field of neuropsychiatry and deserves much needed attention. This article includes case presentations of past drug development programs’ efforts and novel methodological approaches that highlight caveats and potential insights to the future of CNS drug development.
This article is based on proceedings from the “Designing the Right Series of Experiments” session, which was held during the International Society for Clinical Trials Meeting (ISCTM) in Philadelphia, Pennsylvania, September 30 to October 2, 2013.
CASE 1: What we would consider doing differently: Lessons from the mGluR2/3 agonist schizophrenia program
Preface. Pomaglumetad methionil (pomaglumetad) is a potent and highly selective agonist at the metabotropic glutamate 2 and 3 receptors (mGlu2/3R). Here we review the key pivotal steps of the Phase I through III clinical development program at Eli Lilly and Company to assess the efficacy and safety of pomaglumetad in improving the psychopathology of schizophrenia, either as monotherapy in patients with an acute psychotic exacerbation or as an add-on therapy in patients with prominent negative symptoms. Although this program yielded negative primary outcomes, lessons were learned from this significant undertaking that may influence the design and anticipated outcome of future efforts to develop innovative and hopefully more effective drug therapies for schizophrenia.
Preclinical studies. Schizophrenia is believed to be a disorder, in part, due to pathophysiologic dysregulation of glutamatergic neurons, and possibly more so during prodromal and early periods of this disease. Stimulation of mGlu2/3R decreases glutamate release from pyramidal neurons and is hypothesized to equilibrate glutamatergic signaling. Pomaglumetad methionil is the methionine prodrug of the mGlu2/3R agonist LY404039. The prodrug enhances oral bioavailability of the active compound LY404039, which has limited gastrointestinal absorption. Preclinical studies with pomaglumetad suggest this agent to be a potential novel first in class antipsychotic that is devoid of affinity for dopamine (DA) and 5-HT2 receptors. For instance, pomaglumetad dose-dependently reverses the hyperglutamatergic effects of ketamine in key regions of the brain.[1] Ketamine, a NMDA receptor antagonist, produces behavioral hyperactivity in animals that may serve as a model for psychosis in humans. Phencyclidine (PCP or “angel dust”), also a NMDA antagonist and a substance of abuse, produces a psychotic state in humans. Pomaglumetad blocks the effects of PCP in animal models of psychosis as does raclopride, an antagonist at the dopamine D2 receptor, the major biochemical receptor target of traditional antipsychotic agents.[1] Furthermore, the combination of pomaglumetad with risperidone (an atypical antipsychotic with greater 5-HT2:D2 receptor affinity) produces a synergistic inhibition of the conditioned avoidance responses, an animal model of antipsychotic drug activity, greater than either agent alone.[2]
These diverse preclinical data led to early indications that pomaglumetad may be effective as 1) a monotherapy for schizophrenia, and 2) an add on to standard of care antipsychotic treatment for enhanced efficacy (e.g., the adjunctive treatment of persistent prominent negative symptoms). Based upon CSF studies in humans, it was concluded that pomaglumetad 40mg, twice a day, in humans would provide an exposure in the CNS equivalent to that in rat brain which blocks the PCP response. Numerous lines of evidence were thus in favor of pomaglumetad having potential to ameliorate the pathophysiologic aspects of schizophrenia as derived from animal models.
The Phase 2 proof of concept study H8Y-MC-HBBD (HBBD) was carried-out in acutely ill patients with schizophrenia using pomaglumetad 40mg given orally twice a day as monotherapy. This multicenter study (10 sites, single country) demonstrated that pomaglumetad decreased the Positive and Negative Syndrome Scale (PANSS; a rating scale used in schizophrenia) Total Score to similar levels as that of olanzapine in patients with schizophrenia (both treatments were statistically significantly different from placebo).[3] In HBBD, pomaglumetad was well tolerated by patients with a low association to weight gain, extrapyramidal symptoms, hyperprolactinemia, or serious safety or efficacy concern. These positive encouraging results led to a Phase IIb dose-ranging study H8Y-MC-HBBI (HBBI) to assess four doses of pomaglumetad (5mg, 20mg, 40mg, 80mg) to be given orally twice a day in 669 patients from nine countries. However, none of the four doses of pomaglumetad or the olanzapine comparator group in HBBI were subsequently found to separate from placebo resulting in a failed trial that may have been attributable, in part, to a large placebo response.[4]
Subsequent statistical analysis after omitting data from the high placebo response clinical sites (i.e., approximately 25% of the sites) did demonstrate that olanzapine was significantly superior to placebo in the remainder of sites, with pomaglumetad response lying midway in magnitude between the active comparator olanzapine and placebo groups on PANSS-Total score.[4]
Safety concerns. During the HBBI clinical trial, the adverse event of tonic-clonic seizures were observed for the first time in humans with this agent. Prior to HBBI, seizure activity had not been seen in humans treated with pomaglumetad, although seizures were found in rodents. Extensive investigation in non-human primates failed to discern seizures, or seizure-like activity on EEG monitoring, at high exposures for up to one year. After much debate about whether to continue development of pomaglumetad as a treatment for schizophrenia, a long-term (24 week) safety study H8Y-MC-HBBR (HBBR) was designed to treat schizophrenia patients beyond the acute four-week duration limit of studies HBBD and HBBI and to include repeat EEG assessments.[5] Differences in tolerability of pomaglumetad and other standard of care agents (olanzapine, risperidone, and aripiprazole) were compared in the open label 24 week study with time to discontinuation as the primary outcome measure. The pomaglumetad treatment arm (flexible dosed at pomaglumetad 20mg, 40mg, or 80mg, twice a day) in HBBR was well tolerated, and with the longer duration of treatment in HBBR (as compared to the HBBI study), increased patient exposures, and the limited number of any additional seizures, the exposure-adjusted seizure rate on pomaglumetad directionally trended toward the seizure rate reported for currently available antipsychotic agents. Although HBBR was not a placebo-controlled study, pomaglumetad did demonstrate an acute antipsychotic effect on PANSS ratings, which in the initial weeks of treatment was comparable to the standard of care antipsychotic drugs, but which appeared to lessen over longer treatment duration versus the comparator arm.[5] To aid in further understanding the neurobiological correlates and efficacy measures of pomaglumetad, a pharmacogenomic candidate gene analysis approach using patients enrolled in HBBD yielded two alleles of interest: 5-HT2A (rs7330461) and neuregulin 1 receptor (rs10954863).[6] Patients in HBBD that were homozygous for the minor allele of 5-HT2A (T/T) had a robust treatment response to pomaglumetad (i.e., approximately 30-percent reduction on PANSS rating) and homozygotes for the major allele of 5-HT2A (A/A) showed virtually no response to treatment with pomaglumetad. Evaluating similar allelic differences in patients that participated in the HBBR study indicated that patients with the 5-HT2A (T/T) allele also showed a beneficial response to pomaglumetad (not seen in the homozygous major allele group) while this allele may actually confer a reduced response to standard of care agents.[7] It appears that pharmacogenomic data such as these encouraged a tailored therapy strategy for pomaglumetad with potential to be added to a drug development program.
Phase III trial. A path forward to Phase III was based on a better understanding of the seizure incidence associated with pomaglumetad and the potential to develop pomaglumetad as a targeted treatment for an ethnically and pharmacogenetically defined population. A Phase III monotherapy trial H8Y-MC-HBBM (HBBM) was designed to reflect this new pharmacogenomics guidance and utilize a study designed to 1) reduce the large placebo response as encountered in HBBI and 2) lead to a positive treatment trial.
In addition, a proof of concept trial adding on pomaglumetad to a standard of care antipsychotic as an adjunctive treatment for negative symptoms was implemented as a parallel track to registration. Essentially, all patients in HBBM undergoing treatment with pomaglumetad would be analyzed for primary outcome scores and data would also be stratified according to racial (e.g., white male subjects have a greater preponderance of A/A alleles) and pharmacogenomic profiles.[8] Centralized rating and elimination of placebo responders early on were also instituted in the methodology in attempt to reduce the placebo-response rate. Results from HBBM indicated that risperidone was effective in treatment of schizophrenia patients but pomaglumetad failed to separate from placebo at any dose administered, or in the racially defined group from the pharmacogenomics strategy. Adjunctive treatment of pomaglumetad to risperidone, olanzapine, quetiapine or aripiprazole for patients with prominent negative symptoms also did not separate from placebo.[9]
Post-hoc analysis. The negative efficacy results of HBBM led in part to the termination of further development of pomaglumetad. A significant reason for the failure to demonstrate efficacy of pomaglumetad in Phase III may be related to methodological attempts to prevent another failed trial following the HBBI Phase IIb results. HBBM was designed to insure that neuroleptic responsive, by history, patients were recruited in an attempt to facilitate the possible signal detection of treatment response to pomaglumetad. This enrollment enrichment design unfortunately may have been detrimental to the primary outcome results. Limiting enrollment to neuroleptic responsive subjects could have biased the intent to treat population to one with a dopaminergic rather than glutamatergic pathophysiology diathesis based upon their history of dopamine antagonist responsiveness. Therefore, the non-dopamine mechanism of action of pomaglumated may not have had the opportunity to affect response in such patients. A hyperglutamatergic state has been reported to be associated with the early stage of schizophrenia. In a post-hoc analysis, utilizing the HBBM data set as well as an integrated data set of all the placebo controlled pomaglumetad trials, patients early in their disease (10–15% of study patients ?3 years of illness) were identified and found to show a therapeutic response on PANSS-Total scores when treated with pomaglumetad 40mg, orally twice daily. In contrast, patients late in their illness (?10 years) did not receive any benefit to pomaglumetad, suggesting that a possible hyperglutamatergic illness state may be the necessary target for a pomaglumated response.[10] It is also hypothesized that previous exposure to atypical antipsychotics could have led to down-regulation of mGlu2R with a resulting sub-therapeutic response to pomaglumetad, as has been reported to occur in animal models.[11] A post-hoc analysis, once again utilizing the HBBM data set as well as an integrated data set of all the placebo controlled pomaglumetad trials, was thus carried out which dichotomized between patients that were exposed to a subset of typical antipsychotics (identified as predominant dopamine D2 receptor antagonists) or a subset of atypical antipsychotics (identified as prominent 5-HT2A receptor antagonists) in the two years prior to screening visit for any of the pomaglumetad trials in the analysis. This post-hoc analysis did demonstrate that subjects previously exposed to prominent 5-HT2A receptor antagonists (including atypical antipsychotic drugs and other CNS drugs) failed to have significant antipsychotic response to pomaglumetad whereas those patients whose prior antipsychotic exposure was to predominant dopamine D2 antagonists (essentially either haloperidol or fluphenazine) did show a therapeutic response to pomaglumetad.[10]
Lastly, a post-hoc analysis of HBBM demonstrated that non-Hispanic white subjects homozygous for the minor allele of the 5-HT2A receptor snp reported above (T/T allele) once again confirmed PANSS response to pomaglumetad 40mg, when given orally twice a day, as compared to other study cohorts.[12] Although caution should be taken when testing alternative hypotheses through post-hoc evaluations, these results may further our understanding of how a glutamatergic-based therapeutic may require a carefully identified target population rather than the general illness population to show drug efficacy in new drug development clinical trial investigations.
Conclusion. This overview of those specific decision points that led to the progression of pomaglumetad development as well as the ultimate termination of the program may provide lessons learned to help guide alternative decision paths for drug development, certainly for glutamatergic-based compounds with antipsychotic aspirations. Animal models of psychosis may identify valid drug targets but not necessarily identify the appropriate clinical population of the target. Early Phase II clinical trials should be designed to better understand genotypic and phenotypic distinctions between responders and non-responders. Late Phase II trials should be able to conclude on the best targeted approach and optimal drug dose. Phase III trials should be powered appropriately and designed to be confirmatory of prior best possible assumptions. Negative Phase III programs may ultimately provide a useful platform for new hypothesis generation/testing during post hoc examinations. Speculation on the possible reasons(s) for negative efficacy outcomes can be evaluated with the datasets generated from Phase I to III clinical trials. The post-hoc exploratory data analysis of the unique disease-state or disease trait factors that may influence response to pomaglumetad suggest targeted therapeutic strategies that may aid in future clinical trial design.
Lessons learned 1. Spectrum of illness. Only neuroleptic responsive patients in HBBM were included and this could have been a limitation. Early onset patients with schizophrenia may have been more appropriate as a potential hyperglutamatergic mediated disease state may have yielded a greater response to pomaglumetad.
Lessons learned 2. Prior medication effects. Chronic use of neuroleptics may have led to subsensitivity of mGlu2R and prior exposure to 5-HT2 receptor blockade could produce less sensitivity to mGlu2R agonists and lack of treatment response.
Lessons learned 3. Picking the right patient. Individuals with schizophrenia that have the minor allele of the 5-HT2A receptor may be more apt to respond to pomaglumetad.
CASE 2: Delaying the onset of mild cognitive impairment: Epidemiologic trial design incorporating genetic and drug treatment methodology
Preface. Over the last several years, the dementia field has moved toward earlier treatment of Alzheimer’s disease (AD), particularly by incorporating treatments that may have the ability to delay the onset of disease symptoms. This case study will examine the ability of a novel therapeutic strategy to delay the onset of mild cognitive impairment (MCI) due to AD in cognitively normal subjects. In order to conduct a clinical trial of this nature a number of challenges needed to be addressed. First, a potential mechanism to enrich the study population with participants at elevated risk for developing MCI due to AD in the timeframe of the trial (which is five years for this trial) is discussed. Essentially, the use of genetic biomarkers to dictate this enrichment was employed. Second, as subjects entering into the trial will all be cognitively normal, and as expected during the trial some of the subjects will progress to having first indications of abnormal cognition (MCI), use of a battery of tools with sufficient sensitivity to ascertain whether cognition is impaired at the early stages of decline into AD were needed. Third, the therapy that will be administered to delay onset of MCI must have a well-established tolerability profile given the length of this study. As pioglitazone has been used for the treatment of type II diabetes for over a decade, with 22+ million patient years of exposure data, it was felt to meet this criterion. In conclusion, in a fresh approach to AD treatment trials, this study examines a non-amyloid treatment mechanism and targets the role of synaptic energetics via effects on mitochondria in this disease process. This lengthy and innovative clinical trial is overviewed in the context of a design that is powered to address two primary objectives: 1) evaluating the utility of genetic biomarkers to assign risk of developing onset of cognitive decline due to AD over a five-year timeframe and 2) testing the ability of pioglitazone to delay the onset of MCI due to AD in cognitively normal subjects. This program aims to provide new insight into predicting the age related risk for AD as well as evaluating a new therapeutic approach to forestalling the cognitive decline associated with AD, an avenue which seems much more promising than changing the course of illness in the midst of robust symptoms and pathophysiology.
Rationale for using pioglitazone. Peroxisome proliferator-activated receptor gamma (PPARy) agonists, including pioglitazone, modulate a number of pathways implicated in the pathogenesis of AD: energy metabolism, insulin sensitivity, lipid metabolism, amyloid beta homeostasis, and inflammation.[14] PPARy agonists also play critical roles in energy metabolism due to their direct effects on mitochondrial function, biogenesis, and ultimately ATP production in neuronal glucose utilization. Pioglitazone has salutary effects in mouse models of AD and in small sample-sized human studies.[15,16] Furthermore, any pharmaceutical agent used in cognitively normal people must be supported by extensive patient exposure data and be well tolerated if it is to be used in an extended trial design. Given the substantial patient experience and exposure with pioglitazone (22+ million patient years), with this agent known to be well tolerated and relatively safe in a vast majority of patients, part of the rationale with being able to understand its effectiveness at delaying MCI in AD will be carried-out using this drug.
Study design and considerations. The dementia field is moving toward earlier treatment of AD and particularly treatments that delay the onset of the disease. A Phase III clinical trial is currently ongoing by Takeda and Zinfandel Pharmaceuticals to delay the onset of MCI using pioglitazone in a multicenter, double-blind, randomized, placebo-controlled fashion. The trial is poised to enroll approximately 5,800 cognitively normal subjects (male and female), 4,622 subjects deemed to be of high risk for AD (Caucasian), 600 subjects considered at low risk for AD (Caucasian), and 578 non-Caucasian subjects (which includes 60 subjects who are low risk). The current trial design rather more closely resembles that of an epidemiology study recruiting large numbers of healthy elderly subjects while monitoring them for a number of years (5 years), with treatment arms geared to delay the onset of MCI. As instruments that generate outcome measures in AD trials require greater symptomatology to be present than MCI in detecting cognitive changes, the traditional AD trial techniques needed to be modified for MCI, preferably in accordance with a draft United States Food and Drug Administration (FDA) guidance overview detailing clinical research endpoints for prior onset of frank dementia.
Patient selection queries. Particularly with respect to patient selection, FDA does support subject enrichment strategies and guidance on endpoint measures which have appropriately been incorporated into the present detailed initiative. However, there are many questions related to study methodology that need to be addressed during the development of a trial design for delaying MCI rather than treating AD. For instance, in the diagnosis of AD there are cognitive endpoints and functional co-primary outcome measures that need to be established, but MCI by definition does not have a functional decrement that is overtly measurable, thus a different outcome measure without a functional co-primary must be determined. Furthermore, in such a study as this, how is it possible to accomplish a typical Phase II trial goal when treatment trial duration is five years and the sample size prohibits dose finding studies? Rather, could translational medicine and biomarkers be used to guide the dose selection in subjects? Although using biomarkers to enrich the subject population may be possible, can data generated from this approach be used to support outcome measures? As it appears that regulatory agencies at this time are not comfortable with using biomarkers as a primary outcome measure in cognitive impairment, might they be more willing to accept biomarkers as supportive findings? With respect to the latter, would biomarkers that offer support in outcome measures need to be done in all patients? Lastly, does the cognitive demand in MCI translate well in subjects from different countries and how does one carry out a multi-country trial using different languages and cultures when measuring MCI; can one generalize results from a given study population to patients speaking different languages in other countries that are not part of the study? These are just a number of issues, without definitive answers, that were considered prior to launching this trial’s initiative.
Rationale for study design. Given recent failures of AD treatment trials in symptomatic individuals with late MCI and AD, the present study is based on a population subtype that is at the beginning of the AD spectrum. The current study attempts to put forth and evaluate a risk algorithm in cognitively normal patients through use of genetic biomarkers in stratification of risk and treatment response. Specifically, the algorithm will yield a determination of risk in developing MCI due to AD in cognitively normal adults, ages 65 to 83 years, over a five-year time-frame. The initiative will incorporate age, APOE genotype (e2, e3, or e4), and TOMM40-523 genotype [Short (S), Long (L), or Very Long (VL)]. Evaluating a low dose of pioglitazone (relative to doses used in Diabetes) in delaying the onset of MCI due to AD can also add further information regarding subject profiles (Figure 1).
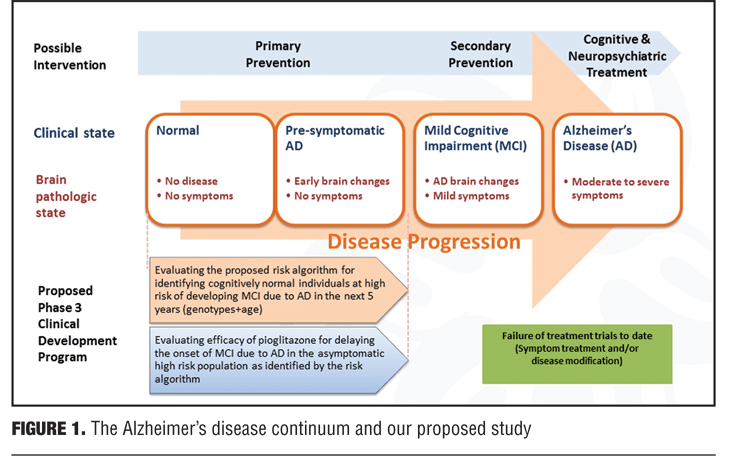
As mentioned, this initiative is an event-based trial with duration of the study as the amount of time needed to achieve 410 events (e.g., detection of MCI) in the high-risk Caucasian group, which is estimated to take approximately four years. A reason for targeting Caucasians in this type of study relates to the prevalence of genetic risk factors for dementia. For instance, the onset of dementia in Japan is approximately 10 years after that of individuals who live in the United States, and the Japanese population has a lower prevalence of APOE4 as a potential contributor to this finding. Given such differences, a number of sites around the world will be participating in this study, such as United States, United Kingdom, Ireland, Italy, Switzerland, Germany, Russia, and Australia to help generalize results from this study to a real-world population.
Enrollment and endpoints. Subjects will be genotyped and placed either into a high-risk or low-risk group according to the biomarkers of relevance. Approximately 2,311 high-risk subjects will be randomized to pioglitazone treatment and an equal number will be randomized to placebo. This randomization is to primarily evaluate the efficacy of the drug treatment while another group consisting of 600 low-risk subjects as determined by the algorithm is to receive placebo. This aspect will be used to qualify the biomarker algorithm as an appropriate risk factor. The low risk group will not be treated using pioglitazone due to the unknown medication efficacy.
Traditional core clinical criteria for MCI endpoints in Phase III studies involve batteries that assess cognitive functioning with performance-based and observational measures. In this study, changes in cognition will be assessed by report from the subject, informant, or clinician (e.g., historical or observed evidence of decline over time), and from objective evidence of impairment in one or more cognitive domains, typically including memory (e.g., formal testing to establish level of cognitive function in multiple domains). For instance, conversion to MCI due to AD would show preservation of independence in functional abilities and criteria consisting of a clinical dementia rating (CDR) scale score of 0.5, AND one of the following: a) fails at least one of the two memory tests in the cognitive test battery; b) fails two or more of the 12 measures in the cognitive test battery representing separate cognitive domains.
There are also many things to rule out in a potential study subject prior to their enrollment, such as vascular, traumatic, and medical causes of cognitive decline. A decision that an individual has an outcome of MCI will be determined through evidence of a longitudinal decline in cognition and continued evidence of cognitive impairment or decline on a six-month follow-up (e.g., 2 consecutive study visits showing impairment). The tests that are used to assess the presence of MCI include five different cognitive domains: episodic memory (California Verbal Learning Test-2nd Edition (CVLT-III); visuospatial memory (Brief Visuospatial Memory Test-Revisited; BVMT-R); 3) executive function (Trail Making Test, Part B); 4) WAIS-III Digit Span Test (backwards span); and 5) language [Multilingual Naming Test (MINT); semantic fluency (animals), Lexical/Phonemic Fluency (FAS); attention (WAIS-III Digit Span Test-forward span; Trail Making Test (Part A), and visuospatial (Clock Drawing Test; Copy of BVMT figures]. An adjudication process will be used in all subjects to make the final determination of MCI. In short, all pertinent data will be shared with an adjudication committee comprising at least three expert clinicians who are blinded to the subject’s risk and treatment assignments. The process will result in one of four possible clinical assessment and subsequent outcomes as measured in the trial design.
This study utilizes a primary outcome of conversion rate to MCI in asymptomatic individuals. Secondary objectives are to the effects of a low dose of pioglitazone on 1) progression of cognitive decline. Safety objectives correspond to long-term safety and tolerability of pioglitazone versus placebo and incidence of treatment-emergent ARIA in cognitively normal elderly patients who received pioglitazone for six months (in a subset of subjects). Exploratory objectives center around the effects of pioglitazone on safety/efficacy of pharmacogenomics to identify genetic loci associated with drug response and/or conversion to MCI due to AD. To this end, effects of pioglitazone on vMRI over time will also be used in the same subset of subjects enrolled in the ARIA substudy.
Retrospective evaluation and optimization of the prognostic characteristics of the biomarker risk algorithm for age of onset of MCI due to AD in non-white subjects will be employed as every six months subjects will undergo assessments. It should be mentioned that routine laboratory tests such as hematology, serum chemistry, and urinalysis will also be taken twice a year.
Phase III extension study. This component is to provide a supportive study for the program and entails that subjects who convert to MCI due to AD be continued on the randomized condition (i.e., pioglitazone or placebo) for at least two years following the study. Based on the feedback from the FDA and EMA, this study could provide important supportive evidence for the novel endpoint measurements of MCI due to AD in the potential marketing application. It is expected that the population will largely comprise of converters from cognitively normal to MCI due to AD during the main Phase III study. The design is essentially a double blind extension phase of the main Phase III study and as mentioned the subjects will simply continue on the same treatment (pioglitazone or placebo). The objective of this extension phase is to evaluate the safety and efficacy of pioglitazone in slowing the progression of cognitive decline in subjects with MCI due to AD. The sample size will be approximately 472 subjects (all converters from high-risk 410 Caucasians + 41 non-Caucasian=451; and low risk=21 subjects) with a number expected to be roughly 316 (67%).
The primary endpoint is the change of cognitive impairment from extension study baseline to at least two years post-study completion (composite cognitive test battery score). Important secondary endpoints in this study will be the number of converters from MCI to AD in each group (Figure 2).
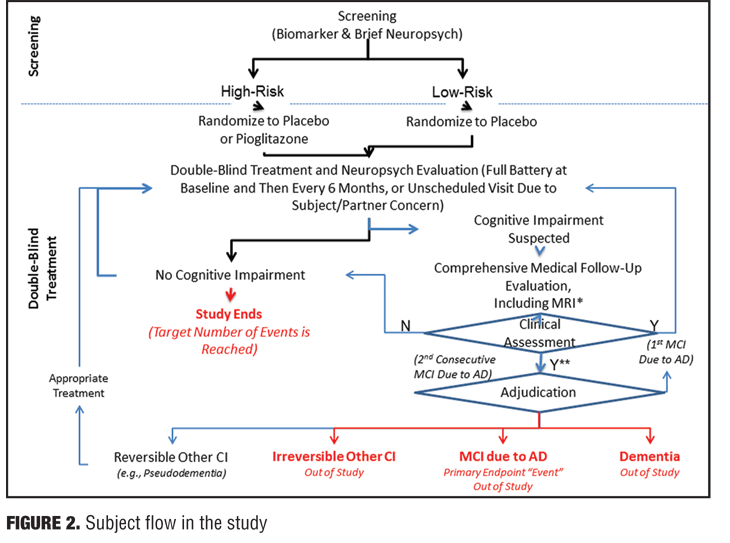
Lessons learned 1. Regulatory agency. Primary endpoint measures can be devised with the help of industry, academics, and government regulatory agencies.
Lessons learned 2. Risk stratification.Using genetic data may help support outcome measures and can be used for risk stratification of enrolling patient populations.
Lessons learned 3. Epidemiological trial design. Longitudinal trial design in prevention of disease in healthy volunteers with drug treatment may require lengthy trial time with prolonged follow-up periods.
CASE 3: Targeting ventral striatal activation and anhedonia in early phase clinical trials of mood and anxiety disorders: The NIMH FAST-MAS perspective
Preface. Despite recent basic science breakthroughs in understanding aspects of the pathophysiology of neuropsychiatric disorders, there is a dearth of new therapeutics in the CNS discovery pipeline. FDA approvals of CNS drugs with novel mechanisms are nearly non-existent and presents a great threat to development of new treatments for psychiatric disease. This lack of innovation is, however, not due to a lack of effort and expenditure in the drug development process. To the contrary, the money invested in failed efforts and the overall costs of successful drug development are rising at an alarming rate. Flaws in the traditional methodology of early phase clinical trials (Phase I and Phase IIa) have been identified as among the most important impediments to the successful development of CNS drugs. Outlined herein is an approach to development of early phase clinical trials that is intended to address some of these critical flaws and is currently being implemented in the NIMH supported program: New Experimental Medicine Studies: Fast-Fail Trials in Mood and Anxiety Spectrum Disorders (FAST-MAS).
Current Phase IIa issues. Phase IIa clinical trial design and methodology is thought to be the biggest contributor to clinical trial problems and is the gate to the most expensive phase of drug development. Phase IIa is a key point where vital information related to a drug candidate is obtained. This includes 1) likelihood of showing an effect with FDA-accepted endpoints, 2) information for designing pivotal trials such as dose, and 3) likelihood of commercial potential. However, Phase IIa trials are frequently underpowered, and the usual clinical endpoints are too variable to test efficacy potential in a Phase IIa study with sufficient power at limited cost. As a result, Phase IIa trial results are vulnerable to bias problems and represent a risky basis for making go/no-go decisions.
Diagnostic issues. Prevailing diagnostic systems are based on consensus and uses clinician observation and patient symptom reports that are phenomenological based. The current diagnostics 1) are not based on science, 2) do not incorporate recent scientific developments, 3) fail to align with neuroscience and genetic findings, 4) are not predictive of treatment response, 5) are unable to capture fundamental underlying mechanisms, and 6) are deficient in aiding in development of new treatment targets to underlying pathophysiological mechanisms. There is reason to believe that current diagnostics may actually be slowing developmental progression of novel treatments and in part contribute to study failure. Specifically, current diagnostics are unlikely to be successful in developing a drug for a condition that 1) does not have a unique pathophysiology and 2) cannot be reliably distinguished from other conditions. It is for these reasons that attention is now being turned to objective neurobiological targets for developing and testing new treatments of interest.
Fundamental misconception. It is becoming increasingly clear that trial failure may stem from not establishing proof of concept (POC) and target engagement for the drug doses studied. Those that go to Phase III prior to establishing POC in terms of efficacy/safety profile in Phase I–II are at risk of a failed trial. Studying dosages that have not been demonstrated to robustly engage the target of interest is problematic. If the results are negative, there is uncertainty as to whether the dose was sufficient. The result is that multiple unnecessary trials are carried out because it is not possible to conclude that engaging the target does not achieve the desired effect with a single negative study.
Another problem is the failure to test specific a priori hypotheses about an agent. The usual focus has been to determine whether a treatment has a therapeutic effect. This results in vulnerability to nonspecific effects and bias. A treatment can appear to have a therapeutic effect for many reasons, some of which are not because the treatment actually improves the condition being treated (e.g., there was an unequal distribution of placebo responders in the treatment groups). As a result, proceeding to Phase III on the basis of a study that only tested the hypothesis of whether there is evidence of a beneficial effect is a setup for failing to replicate.
Proposed solution using quick-win/fast-fail approach. The focus is on designing early phase studies that achieve POC early through employment of biomarkers and surrogate endpoints. Once a dose is established and robustly engages the target, studies can be carried out that rigorously test the hypothesis of whether engaging the target achieves a hypothesized effect as a means of establishing POC. Objective biomarkers are likely closer to pathophysiology and therapeutic mechanism than clinical endpoints while possessing less variability and greater power. As a result, a smaller trial with less variability and cost can be carried out to determine POC. Furthermore, since the vast majority of drug candidates fail, this methodology provides a means to allow treatments that should not be further developed to fail more quickly and at less cost.
This, however, requires a shift of R&D investment from later to earlier stages of drug development. It is proposed that designing Phase I/IIa studies to definitively indicate therapeutic potential is critical to success. Such studies must establish POC using biomarkers/surrogate endpoints early on as a basis for determining whether a treatment that engages a target achieves the desired effect on physiology or objectively measureable behavior.
This strategy is not only less costly overall, it also makes it more likely that Phase III studies that are carried out will be more likely to replicate, as some of the sources of bias that create false positive Phase IIa results will have been eliminated. However, for this type of work to be possible, it is necessary to develop reliable biomarkers of both efficacy and safety for a variety of conditions, and this will require a substantial effort.
Research Domain Criteria (RDoC) framework. RDoC is a new framework that incorporates reseach on pathophysiology, including emerging findings from genomics/neuroscience research.
As RDoC is based on research findings, it is more likely to be useful than prior approaches for identifying promising new treatment targets, detecting key subgroups, and developing valid outcome assessments. It is also more likely to achieve correspondence between animal models and human conditions of interest. RDoC-defined “constructs” are key dimensions of function that are important for neuropsychiatric function. They are defined by research studies related to that dimension of function, and they are subject to continual refinement based on advances in science. This includes studies identifying relevant associated genes, molecules, cells, circuits, physiology, behavior, self-report, and experimental paradigms. Related constructs are grouped into five major domains of functioning: 1) Negative Valence Systems (i.e., systems for aversive motivation), 2) Positive Valence Systems, 3) Cognitive Systems, 4) Systems for Social Processes, and 5) Arousal Regulatory Systems.
Recently, the NIMH established networks for carrying out early phase trials of molecules engaging promising new targets using the FAST-FAIL methodology described earlier and working within the RDoC framework. These networks represent unique NIMH, private, and academic partnerships are meant to 1) provide a path for promising compounds that are currently not being developed, 2) establish a new standard for early phase drug development that will improve the cost/benefit of CNS drug development and re-kindle pharma interest, and 3) improve our understanding of mechanisms of psychopathology, treatments to psychiatric disorders, and optimal trial methodologies. The NIMH has recently awarded grants in three areas: 1) Mood and Anxiety Spectrum Disorders (FAST-MAS; a grant awarded to Duke; PI – Krystal), 2) Psychotic Spectrum Disorders (FAST-PS; grant awarded to Columbia; PI – Lieberman), and 3) Autism Spectrum Disorders (FAST-AS; grant awarded to UCLA – PI – McCraken).
These efforts involve a number of steps including choosing a neurobiologic target that is promising but yet to be fully evaluated; finding a molecule that engages that target; determining a dose of the molecule that robustly engages that target; identify an appropriate RDoC construct relevant to the target of interest; select outcome measures that reflect that construct and that will allow an assessment of whether engaging the target achieves the desired effect on the brain; identifying the relevant patient population and determining how they will be selected; and estimating the number of subjects needed for the trial. Perhaps the most important step is selecting the target to study. This has involved working with a committee including NIMH program offices and consultants to identify the most promising targets to study and how to study them. Once this has been achieved, the next key step is to design trials based on the principles outlined earlier to optimize the capacity to make a definitive go/no-go decision at the end of Phase IIa. It should be emphasized that go/no-go decisions will be made based on whether engaging the target achieves a hypothesized effect, such as achieving a change in a particular brain circuit, and not on whether there is a significant effect on a clinical scale. If the drug does not engage the circuitry it would not make sense to move forward even if there is a therapeutic effect on a clinical or self-report endpoint. This would be another likely set up for a failed Phase III trial.
Qualification process for the FAST-MAS target selection process. First, there should be promising preclinical/clinical data that indicate the potential of a given target. There also has to be available means of assessing target engagement, and an available molecule for testing the target hypothesis. The drug must also be far along enough that an Investigational New Drug application (IND) exists or it is IND-ready, as time is limited. The molecule must not be too far along in development such that the promise of the target (or lack thereof) has already been established or such that trials are being carried out that are redundant with likely FAST-MAS trials. Furthermore, a target must have an associated molecule that does not have prohibitive adverse effects or a problematic toxicological profile. In terms of being able to assess target engagement, it is necessary to have means to determine the degree to which a given dose engages the pharmacologic target of interest (e.g. antagonism of the 5HT2A receptor). It is also necessary to have means to test whether engaging the target has the hypothesized effect (e.g., achieves a change in a brain circuit or an objectively measureable aspect of behavior).
Process of target selection. As part of the FAST-MAS effort, an exhaustive search was carried out that led to identifying a target of interest where there was a molecule available that engaged that target and that was at an appropriate point in development. Means of establishing a dose that robustly engages the target with PET was an an important factor in choosing the molecule to study. The same was true for having means of establishing POC in terms of an effect on brain circuits.
In the case of FAST-MAS, where we are studying a molecule that is believed to affect reward function, the relevant test of whether there is an effect on the brain is a determination of whether treatment affects reward-related circuitry.
Choice of RDoC constructs(s) challenge. The first question that one needs to ask is “Where in RDoC is the functional domain that I want to study?” For instance, our interest was in studying the inability to experience pleasure, referred to as anhedonia (a core symptom of major depression), which cuts across traditional diagnoses as anhedonia is also seen in those with anxiety disorders and schizophrenia. As a result, we sought to identify RDoC constructs related to anhedonia and identify the associated circuitry, behavior test, and clinical scales to employ. The relevant RDoC domain for anhedonia is Positive Valence Systems. There are a number of relevant RDoC constructs related to reward, such as Reward Valuation, Expending Effort for Reward, Reward Prediction/Expectancy, Reward Responsibility, and Effect of Reward on Learning. These constructs reflect current best understanding of relevant neurobiology and tools for assessment of outcomes. A number of the relevant circuits and measures overlap to a degree across constructs of interest. We chose to employ task-related fMRI using the monetary incentive delay task to determine whether engaging our target had the hypothesized effect on neural circuitry (increase in ventral striatal activity) as a means of establishing proof of concept. This task is known to change in response to antidepressant treatment in depressed subjects.
Outcomes are reverse of traditional approach. A circuit measure serves as the primary outcome measure for our study. Key secondary measures are behavioral intermediate phenotype assessments (more closely linked to neural circuitry than clinical outcome, but also linked to clinical outcome). The probabilistic reward task assesses the capacity to learn based on reward. We also employed a clinical outcome scale that has been demonstrated to be sensitive to treatments—the Snaith-Hamilton Pleasure Scale (SHAPS).
Exploratory investigations will include 1) additional circuit measures (i.e., OEEG measure of cingulate activity), 2) additional behavioral measure (i.e., effort expenditure for reward tasks) assesses the degree to which one is motivated by reward as demonstrated by effort, 3) additional self-report scale (i.e., Temporal Experience of Pleasure Scale [TEPS]). We also included the HAM-A and HAM-D to allow linking this study to prior DSM-based work. We considered using the Snaith-Hamilton scale for pleasure as the primary outcome measure. However, it seemed likely that this would result in our study being significantly underpowered and risky. Instead, a circuit outcome was used as primary outcome and as a basis for go/no-go decisions. If the circuitry is not engaged, it would not make sense to move forward in development with our compound, even if there is a therapeutic effect on a clinical or self-report endpoint. This would likely be a setup for a failed Phase III trial.
Choice of subject population. One novel aspect of our study design is to employ the Snaith-Hamilton Pleasure scale as a screening tool to identify subjects with anhedonia. We would have liked to use our measure of ventral strial circuitry activity level for this purpose; however, the work completed to date provided an insufficient basis for doing so. It is important to note that we are also requiring people to have a DSM-based diagnosis of a mood or anxiety disorder in addition to meeting anhedonia entry criteria. This ensures that we will be studying subjects with reward-related difficulty who have a mood and anxiety spectrum disorder. However, because anhedonia is a diagnostic symptom of depression, we felt it important to take steps to ensure that our study has cross-diagnostic relevance in keeping with the RDoC approach. Based on input from our consultants, it was concluded that our options were to demonstrate that treatment improves anhedonia but NOT a depression scale (HAM-D/MADRS) or that treatment improves anhedonia across DSM diagnoses in a population where some individuals do not meet criteria for major depression. We took that latter approach and decided to include a subset of subjects who met DSM criteria for an anxiety disorder and did not meet criteria for major depression.
General limitations/risk of fast-fail we faced. There are challenges in determining number of subjects for FAST-MAS studies. We decided to carry out a study that is powered to detect an effect size of 0.5 in our primary outcome measure: ventral striatal activation during the monetary incentive delay task based on fMRI With this stated, the capacity to estimate effect size on the primary outcome measure was limited by there being very few treatment studies having been carried out with monetary incentive delay task fMRI.
Our experience has illustrated that carrying out studies using the FAST-MAS approach will require further developing means of establishing target engagement and establishing POC. We currently lack the means to do this for many RDoC constructs and potential targets. Further, the FAST-MAS strategy cannot prevent Phase III failures due to relatively uncommon unanticipated adverse effects due to studying a small number of people, but should be a more efficient and more cost savings approach.
Lessons learned 1. Target selection. Choosing the appropriate target requires a great deal of time and consideration.
Lessons learned 2. Outcome measures. There is a great need to develop a means to establish target engagement and to assess whether engaging a target has the hypothesized effect on brain/behavior.
Lessons learned 3. Logistics. Coordinating different aspects of this initiative required partnerships between government, academic, and industry.
CASE 4: The pharmacological characteristics of a therapeutically active dose of two first-in-class-drugs: Amorexant (ORX1/2 antagonist) and Rimonabant (CB1-antagonist)
Preface. By definition, first in class drugs lack registered congeners that can be used to benchmark the activity profiles, in such cases, dose selection for clinical trials is usually based on an assessment of preclinical safety and efficacy data, and on pharmacokinetics and tolerability in healthy subjects. Unfortunately, this frequently leads to disappointing clinical trial outcomes, and few truly innovative CNS-active drugs have made it to the clinic in the past decade. Moreover, a large proportion of drugs that are registered become subject to reductions of recommended doses after launch, or are withdrawn from the market. Even though validated pharmacological endpoints are often lacking for new drugs, it can still be useful to characterize the pharmacologic effects of first in class drugs in healthy individuals. This can provide important information about the various dose- or concentration-effect relationships for potentially desirable or detrimental effects of the new compound. This offers the possibility to rationally select a dose with specific pharmacologic activity, and to cautiously escalate the dose along the concentration effect curve during testing. An approach such as this may prevent unexpected clinical outcomes, which otherwise may occur if the pharmacologic activity of a traditionally selected dose becomes outside of the optimal therapeutic-safety window. Pharmacologically guided dose selection can also increase confidence that the drug is not overdosed. Two examples of this approach are provided. For the first dual orexin antagonist, pharmacologically guided dose selection was successfully used to identify a minimal sleep-promoting dose and to avoid potential narcolepsy-like effects. The second example is for a cannabinoid antagonist, where pharmacologically guided dose estimation was only implemented after rimonabant, the first compound in this class, showed unexpected psychiatric adverse effects following launch.
A focus on pharmacology. Potential causes of highly selective, innovative drugs not progressing favorably during the drug development process may relate to not getting the pharmacology right. Arrowsmith[17] essentially details that failure of compounds in Phase II trials was largely attributed to a lack of efficacy. It would be expected that failures in Phase II trials would reduce the risk of efficacy failure in Phase III, but, unexpectedly, two-thirds of failed trials in late-stage development are still caused by lack of efficacy.[18] The lack of efficacy is not due to lack of understanding of the disease, per se, but of more specific factors, such as insufficient brain penetration or target engagement or increased variability of these factors in a clinical Phase III population due to interactions with disease or co-medication. In a sense, “failed efficacy” reflects the inability to understand Phase II trial results and to cultivate effective clinical trial initiatives from these results for later phase studies. Limitations, such as a lack of understanding of how drug targets impact the pathophysiology of the disease, the variability of the disease in the population, and dose efficacy trials only being defined in select populations, could also lead to drug development failures.[19] It seems that after performing Phase I trials, target engagement is almost completely forgotten and it is unknown if the drug still has the effect in the clinical population or in later phase trials. Take, for instance, the 25 drugs that were withdrawn after launch out of 275 drugs that were registered within the same period between the years 2006 and 2011—a third of these was later attributed to adverse events that were predictable and largely reflective of dose-related issues and lack of target engagement. From 1980 to 2000, dose reductions occurred in 27 percent of all new FDA registrations of CNS-active drugs, and 79 percent were due specifically to safety-related effects. This pattern occurred three times more often between the 1995 and 1999 than between the years 1980 and 1985.[20] Most dose reductions were pharmacologically based and highlights how getting the pharmacology right in early stages of development before a drug goes into patients can save more money and lead to improvements in clinical trial outcomes.
Safety windows and therapeutic efficacy. Increasing the dose until the maximum tolerated dose is reached is difficult with drugs that have a small therapeutic window. Mainstay agents in psychiatry have small margins of safety and efficacy (such as older agents), and investigators are increasingly trying to develop drugs that have a wide margin of safety. To this end, selective or allosteric modulators, rather than direct or indirect agonists/antagonists, may be able to exhibit more favorable safety profiles. Drugs with larger therapeutic safety margins in healthy volunteers are used to measure efficacy, and often the aim is for the choosing the highest dose range with greatest chance of a therapeutic effect being reached. This approach can kill many drugs that have U-shaped dose curves and likely leads to dose reductions after launch or side-effects in patients that were unnecessary. So what needs to be done to get the dose right? First, it is important to consider that highly selective drug pharmacology that can cause efficacy is the same pharmacology that can also cause other events, and second, both of these can be measured in healthy volunteers to provide the therapeutic range measured effects.
There is a range of different pharmacological endpoints with good PK/PD relationships that can exist, and these different effects will be in the therapeutic range. Screening new drugs with a series of different biomarkers that are sensitive to various agents will be able to produce a profile of drug effects that may be more encompassing than just single measures.
These types of investigations can inform about the active concentration range that will drive therapeutic effect. Furthermore, pharmacology and efficacy thereof can vary quite a bit for different classes of antipsychotics. We know that D2 receptor occupancy needs to be in the range of 60 to 80 percent for antipsychotics, and for benzodiazepines it is between 5 to 30 percent. Larger dose ranges for compounds of similar modes of action can occur, and knowing how much target engagement is needed for a therapeutic effect is difficult to determine.
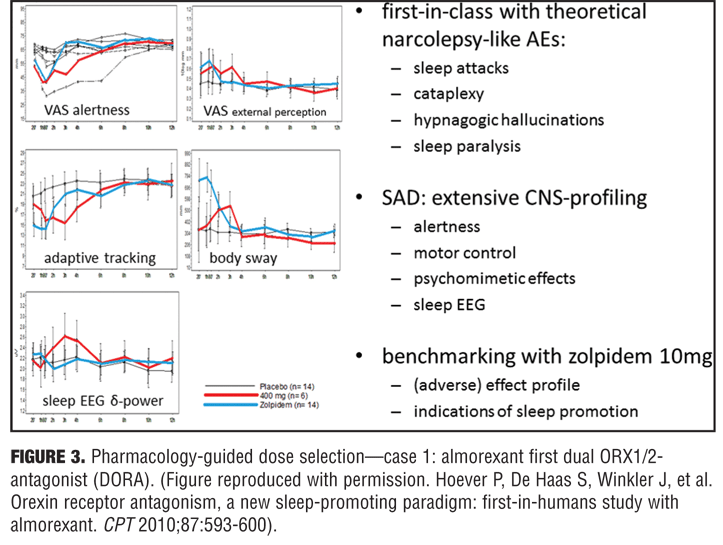
Optimizing drug action. Pharmacology-guided dose selection for first in class CNS active drugs: Example 1—the dual orexin antagonist almorexant. Orexin is deficient in patients with narcolepsy, and patients with narcolepsy have sleep attacks. It is the latter that may be beneficial to induce in some individuals with sleep problems (e.g., insomnia) with almorexant, an oxerin antagonist, but not the associated sleep paralysis, hypnogogic hallucinations, or cataplexy that is found associated in patients with narcolepsy. No one knows how large of a therapeutic window for almorexant would be needed given that it is a first in class agent. A pharmacologic approach was taken that started at the low end of the concentration window to determine the safety window and was progressive in dose escalation until a detrimental effect was observed. Dosages before this safety concern result would ensure that a desired effect was not missed and provide confidence in the positive results if demonstrated with the agent on sleep. A positive control was also used to benchmark the effect by inclusion of zolpidem 10mg. Effects of 400mg of almorexant and 10mg of zolpidem induced a strong reduction in visual analog scales (VAS) of alertness, but at this almorexant dose the effects were much lower on body sway and potentially indicated that this drug may have a reduced propensity for falls.[21] Reduction in delta waves are thought to be an indicator of a lack of deep sleep and through PK/PD modeling of almorexant it was determined that during sleep subjects showed greater delta activity and deeper sleep (i.e., a property that zolpidem does not have). Inferred from these results that this first in-man study with almorexant could provide an indication of the pharmacology and PK/PD modeling needed to determined what the clinically effective dose would be, seemingly below 400mg and in the region of 100 to 200mg, which would be at the low therapeutic range (i.e., similar to that of an effect with zolpidem 5mg). In the end, this was deemed to be a successful program, as a 50mg dose of almorexant was not an effective sleep aid and 100 to 200mg exhibited dose-dependent effects in patients with insomnia. The 200mg dose did exhibit significant safety concerns and the compound was later withdrawn for side-effects that were not associated with the drug effect on sleep parameters and not being deemed to be a class problem. Merck Corporation has recently received approval for Suvorexan (another orexin antagonist) using a similar approach (Figure 3).
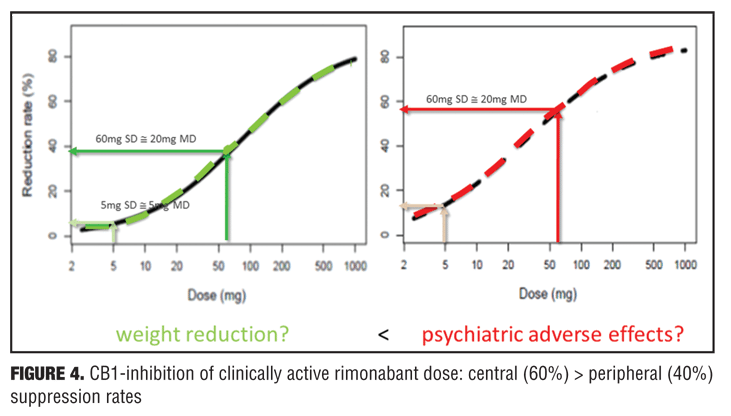
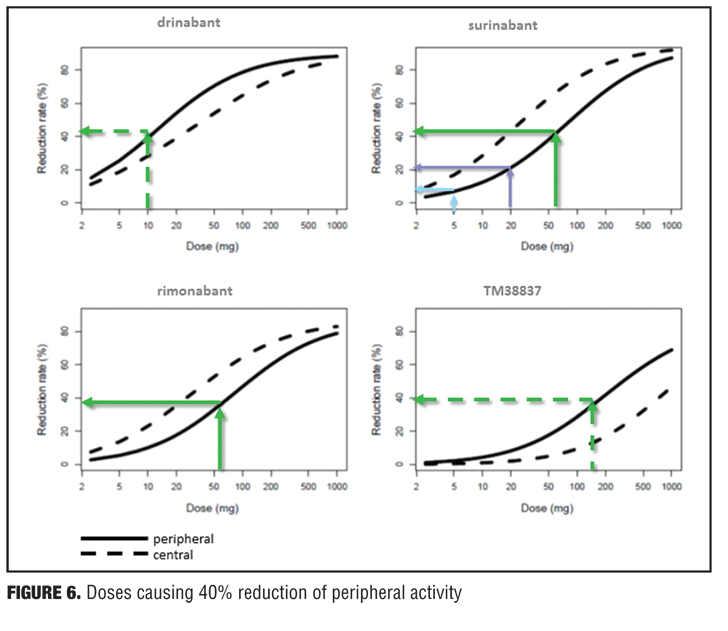
Example 2—Cannabinoid CB1 antagonist rimonabant and surinabant. Unlike the abovementioned case with almorexant, this is more of a postmortem case as to try to understand what went wrong. Specifically, the reports of four suicides in the rimonabant group versus one in the placebo group initially led to drug termination. There were many other side effects in the psychiatric group leading to halting of this drug during clinical development, which contributed to the entire drug class being taken with it. Rimonabant is a drug registered by Sanofi-Aventis and had not undergone pharmacodynamic evaluation in healthy volunteers as had the drug in the previous example. However, the drug was capable of reducing the effects of THC and represented a starting point for devising studies in the evaluation of surinabant. It was decided to first isolate THC from cannabis and then devise an inhalation method to determine peripheral/central pharmacologic activity using PK/PD analysis with relevance of these measures to be used later in clinical trials.[22] As it was not known how much suppression in THC effects was needed according to these models, essentially investigators had to wait for clinical trial results with rimonabant to be able to reverse model back. Interestingly, plasma concentrations of rimonabant disappears rapidly but CNS effects remained prolonged. Heart rate effects did not accumulate to additional dosages of drug, and this in fact was deemed to be a peripheral CB1 receptor effect versus impact on central pharmacologic activity.[23] Given the ability to parse peripheral and central effects out with this agent, this rational approach was then undertaken with the follow-up agent surinabant. Fittingly, there is a dose-related reduction in heart rate with surinabant, and if a PK/PD model is created, it was hoped that what would be the highest dose that one could achieve a 10-percent reduction in heart rate would be the effective dose. This level of inquiry would still, however, not lead to anything related to what would be the dose needed for weight reduction (a primary endpoint of the study drug). A dose-related reduction of feeling high from THC was only appreciated with ribonabant 60mg, and caused only a 60-percent reduction in this subjective effect. The types of side effects encountered with surinabant also occurred within the same dose range as that for ribonabant. There was indeed a dose-related weight reduction in people who quit smoking, and 20mg of ribonabant appeared similar to the 10mg of surinabant when tested, which showed levels of THC suppression at 40 percent peripheral and 60 percent centrally. So the question was “Which measure is needed to see the desired weight reduction effect?” Many thought that after these studies, lower doses of drug related to the peripheral effect is what was needed, and then came along drinabant. Drinabant is a cannabinoid analog more peripherally restrictive, and 7TM is also restricted in central penetration and considered to be clinically effective in weight reduction. But these latter drugs never underwent clinical trials. In short, it was felt that much lower levels of central penetration will be associated with reduced psychiatric side effects while still maintaining the efficacy of weight reduction outcomes
(Figures 4–6).

Lesson 1. Human pharmacology. For selective drugs, pharmacodynamics using active concentrations in healthy subjects are often closely related to therapeutic levels. Pharmacokinetic-pharmacodynamic relationships are an important aspect of proof of concept drug development.
Lesson 2. Objective biomarkers. Drugs can potentially play a role in the RDoC frameworks. Pharmacology guided dose escalation allows maximization of therapeutic window. This may avoid adverse events associated with unnecessarily high doses. Increased confidence in dose optimization is the result.
DISCUSSION
In all of the presented cases, a tremendous emphasis was placed on choosing the correct population to enroll into the treatment trial. In Case 1 with pomaglumated, enrolling subjects with treatment-resistant schizophrenia appeared to not be the most optimal cohort as post-hoc analyses indicated that early onset and atypical antipsychotic naïve subjects with schizophrenia may have been the most appropriate cohort to test with this compound. In Case 2, using genetic data to stratify subjects based on risk of MCI due to AD is essential for enrollment in clinical epidemiologic type trial designs. In Case 3, the ability to evaluate neurobiologic changes to drug effect in individuals with symptom of anhedonia, which can cut across numerous psychiatric illness, will be important in assessing if the drug can treat individuals with this presentation. In Case 4, it may have been that testing compounds in healthy volunteers with objective biomarker endpoints would have dictated a more favorable pharmacologic dose portfolio and drug information when it came to testing in certain clinical populations. For these case presentations, choosing the appropriate patient population, as well as having objective biomarkers that direct therapeutic evaluations is needed. Multicenter trials are the mainstay in gaining FDA approval of a therapeutic for a psychiatric disorder indication, it does appear to add considerable variance to data collection and generation of results that may not be in keeping with ability of traditional designs to separate drug effects from placebo. By instituting neurobiologic primary endpoints with symptom rating scales as detailed in Case 3, the ability to decrease variance could be potentially accomplished, but methodology across all sites would still have to be standardized. It appears that more rigorous testing and planning at early stages using neuropharmacologic objective biomarkers could lead to better Phase III study design and dose selection which favors successful treatment trial outcomes.
Concluding Remarks
Changing the way in which individuals are classified and selected for enrollment into drug development initiatives, with use of endpoints that are objective, could provide great benefits to clinical trial programs (Figure 7). Spending more time categorizing target engagement with phenotypic relevance and employing biomarkers that are objective benchmarks for dose-selection and safety parameters could result in fewer negative and failed trials. A decrease in the overall cost of programs by shifting focus at an appropriate time dictated by results generated at the earlier stages of development is prudent. By doing so, post-hoc analysis may yield to detailing a more homogenous patient population by use of genetics and objective endpoints that could lead to less variance in positive outcome measures when re-testing the compound. Characterizing effective dose ranges and safety profiles in humans prior to beginning these investigations may also lead to great benefits with agents making it to market and staying there.
Acknowledgments
We thank Ashish Duggar, PhD, Judith Dunn, PhD, and Richard Keefe, PhD, for their contributing comments and manuscript edits.
References
1. Fell MJ, Perry KW, Falcone JF, et al. In vitro and in vivo evidence for a lack of interaction with dopamine D2 receptors by the metabotropic glutamate 2/3 receptor agonists 1S,2S,5R,6S-2-aminobicyclo[3.1.0]hexane-2,6-bicaroxylate monohydrate (LY354740) and (-)-2-Oxa-4-aminobicyclo[3.1.0] Hexane-4,6-dicarboxylic acid (LY379268). J Pharmacol Exp Ther. 2009;331(3):1126–1136.
2. McKinzie, DL, Fell, MJ, Johnson, BG, et al. Synergistic interactions of the mGlu2/3 receptor agonist LY404039 with antipsychotic agents in behavioral and neurochemical animal models predictive of antipsychotic efficacy. Schizophr Bull. 2011;37:110.
3. Patil ST, Zhang L, Martenyi F, et al. Activation of mGlu2/3 receptors as a new approach to treat schizophrenia: a randomized Phase 2 clinical trial. Nat Med. 2007;13(9):1102–1107.
4. Kinon BJ, Zhang L, Millen BA, et al; HBBI Study Group. A multicenter, inpatient, phase 2, double-blind, placebo-controlled dose-ranging study of LY2140023 monohydrate in patients with DSM-IV schizophrenia. J Clin Psychopharmacol. 2011;31(3):349–355.
5. Adams DH, Kinon BJ, Baygani S, et al. A long-term, phase 2, multicenter, randomized, open-label, comparative safety study of pomaglumetad methionil (LY2140023 monohydrate) versus atypical antipsychotic standard of care in patients with schizophrenia. BMC Psychiatry. 2013;13:143.
6. Liu W, Downing AC, Munsie LM, et al. Pharmacogenetic analysis of the mGlu2/3 agonist LY2140023 monohydrate in the treatment of schizophrenia. Pharmacogenomics J. 2012;12(3):246–254.
7. Nisenbaum LK, Zhao F, Downing AM, et al. Confirmation of association between genetic markers in 5HTR2A and response to mGLU2/3 agonist LY2140023 monohydrate in schizophrenia. Eur Neuropsychopharm. 2011; 21(Suppl 3):S233–S244.
8. Downing AM, Kinon BJ, Millen BA, et al. A double-blind, placebo-controlled comparator study of LY2140023 monohydrate in patients with schizophrenia. BMC Psychiatry. 2014;14:351 doi:10.1186/s12888-014-0351-3.
9. Stauffer VL, Millen BA, Andersen S, et al. Pomaglumetad methionil: no significant difference as an adjunctive treatment for patients with prominent negative symptoms of schizophrenia compared to placebo. Schizophr Res. 2013;150(2–3):434–441.
10. Kinon BJ, Millen BA, Zhang L, McKinzie DL. Exploratory analysis for a targeted patient population responsive to the metabotropic glutamate 2/3 receptor agonist pomaglumetad methionil in schizophrenia. Biol Psychiatry March 2015. In press.
11. Kurita M, Holloway T, García-Bea A, et al. HDAC2 regulates atypical antipsychotic responses through the modulation of mGlu2 promoter activity. Nat Neurosci. 2012;(9):1245–1254.
12. Nisenbaum LK, Millen BA, Zhao F, et al. LY2140023 monohydrate in the treatment of patients with schizophrenia: pharmacogenetic analysis within a clinical trial assessing efficacy in treating acutely ill patients. Schizophr Bull. 2013;39 (Suppl 1):S105.
13. Landreth G, Jiang Q, Mandrekar S, Heneka M. PPARgamma agonists as therapeutics for the treatment of Alzheimer’s disease. Neurotherapeutics. 2008;5(3):481–489.
14. Sato T, Hanyu H, Hirao K, Kanetaka H, Sakurai H, Iwamoto T. Efficacy of PPAR-? agonist pioglitazone in mild Alzheimer disease. Neurobiol Aging. 2011;32(9):1626–1633.
15. Nicolakakis N, Hamel E. The Nuclear Receptor PPARgamma as a Therapeutic Target for Cerebrovascular and Brain Dysfunction in Alzheimer’s Disease. Front Aging Neurosci. 2010;2. pii: 21.
16. Insel TR. The NIMH Research Domain Criteria (RDoC) Project: precision medicine for psychiatry. Am J Psychiatry. 2014;171(4):395–397. doi: 10.1176/appi.ajp.2014.
17. Arrowsmith J. Trial watch: Phase II failures: 2008-2010. Nat Rev Drug Discov. 2011;10(5):328–329.
18. Arrowsmith J. Trial watch: Phase III and submission failures: 2007-2010. Nat Rev Drug Discov. 2011;10(2):87.
19. Cohen AF. Developing drug prototypes: pharmacology replaces safety and tolerability? Nat Rev Drug Discov. 2010;9(11):856–865.
20. Cross J, Lee H, Westelinck A, Nelson J, Grudzinskas C, Peck C. Postmarketing drug dosage changes of 499 FDA-approved new molecular entities, 1980-1999. Pharmacoepidemiol Drug Saf. 2002;11(6):439–446.
21. Hoever P, de Haas S, Winkler J, et al. Orexin receptor antagonism, a new sleep-promoting paradigm: an ascending single-dose study with almorexant. Clin Pharmacol Ther. 2010;87(5):593–600.
22. Klumpers LE, Roy C, Ferron G, et al. Surinabant, a selective CB1 antagonist, inhibits THC-induced central nervous system and heart rate effects in humans. Br J Clin Pharmacol. 2013;76:65–77.
23. Ferrona G, Klumpers L, Van Gerven J, Roy C. PK and PK/PD modeling of CB1 blocker antagonism of THC induced CNS and Heart Rate effects. PAGE Poster 2010.