
by Jared W. Young, PhD; William Z. Potter, MD, PhD; Steve Riley, PharmD, PhD; Geert J. Groeneveld, MD, PhD; Bruce J. Kinon, MD; Mike F. Egan, MD; and Douglas E. Feltner, MD
Dr. Young is with the Department of Psychiatry, University of California San Diego, La Jolla, California, and Research Service, VA San Diego Healthcare System, San Diego, California; Dr. Potter is with the National Institute of Mental Health, Rockville, Maryland; Dr. Riley is with the Department of Clinical Pharmacology, Global Innovative Pharma Business, Pfizer, Inc., Groton, Connecticut; Dr. Groeneveld is with Center for Human Drug Research, The Netherlands; Dr. Kinon is with Lundbeck LLC, Deerfield, Illinois (Dr. Kinon was with Eli Lilly and Company, Indianapolis, Indiana, when this material was presented); Dr. Egan is with Clinical Neuroscience, Merck & Co, Inc, North Wales, Pennsylvania; and Dr. Feltner is with AbbVie Inc. Pharmaceuticals, Chicago, Illinois.
Innov Clin Neurosci 2015;12(1–2 Suppl A):5S–10S.
Funding: No funding was provided for the preparation of this article.
Financial disclosures: In the past three years, Dr. Young’s work has been funded by NIDA and NIMH, as well as the US Veteran’s Administration VISN 22 Mental Illness, Research, Education, and Clinical Center, Cerca Insights, Lundbeck Ltd, and Omeros; Dr. Young has also received consulting compensation for Amgen. Dr. Potter reports consulting fees for Amgen, Lilly, Ironside, Takeda, and Taisho. Dr. Riley is an employee of Pfizer, Inc. Dr. Groeneveld reports no financial disclosures relevant to the content of this article. Dr. Kinon is an employee of Lundbeck LLC (was an employee of Eli Lilly and Co. when this material was presented). Dr. Egan is an employee of Merck. Dr. Feltner is an employee of AbbVie Pharmaceuticals.
Key words: Drug discovery, pharmacology, FDA, pharmacokinetics, decision-making, proof of concept, target rejection
Abstract: For decades, there has been a distinct disconnect translating a compound’s effects from basic neuroscience into clinical efficacy. This disconnect has not only been in terms of generating approved compounds, but also in rejecting targets. During the drug discovery process there are key points to be adhered to that would strengthen the likelihood of a compound being translated to the clinic. These points include 1) the importance of translational pharmacology whereby preclinical pharmacological data should predict clinical efficacy; 2) rigorous early phase drug evaluation to enhance early go/no-go decision-making; 3) using exposure response modeling to predict drug efficacy during proof-of-concept trials; 4) designing and conducting the appropriate proof-of-concept study; and 5) optimizing Phase II studies to set the stage for success in Phase III trials. These topics were covered in The International Society for CNS Clinical Trials and Methodology (ISCTM) Autumn 2013 meeting on the topic of translational and early development strategies and tools led by Drs. Potter and Feltner. This report comprises a review of those proceedings with a concluding summary to advance future clinical trials.
Introduction
Within translational medicine, there remains a critical need for the successful translation of the effects of compounds developed from basic neuroscience to their effects in the clinic. This gap in translation is not only in terms of generating United States Food Drug Administration (FDA) approval, thus validating the target in the marketplace, but also in terms of difficulty in target rejection—giving up on a target despite clinical evidence. For example, in recent years there have been more than 75 targets for antidepressants, and while three have been validated, only three have been rejected, leaving many still being pursued because of lack of ability to finalize rejecting a target. For psychosis, including cognition in schizophrenia, a similar pattern emerges where, in more than 50 targets, only one has been validated and only three have been rejected. For Alzheimer’s disease, this situation is even more pronounced: in more than 150 targets, only one has been validated and none have been rejected. One goal in drug development research should be to develop innovative methodology for fully rejecting targets. In order to improve target validation and rejection, a greater focus is required on the Target Validation Space (i.e., preclinical research through to FDA/Investigative New Drug application (IND) Phases I through III.
A target can be thought of as being a molecular structure/site in the brain or a particular electrical current. Research Domain Criteria (RDoC) is a novel way that the National Institute for Mental Health (NIMH) has devised whereby a target can be exactly as described above and a site is targeted that may underlie a symptom without necessarily being associated with a disease. RDoC avoids using the fourth or fifth editions of the Diagnostic and Statistical Manual of Mental Disorders (DSM-IV, DSM 5) and instead focuses on particular systems underlying specific behaviors. These systems include Negative Valence Systems, positive valence systems, cognitive systems, social systems, arousal, and modulatory systems.[1] A new program (Fast fail trials) has also been designed to begin targeting such systems, especially examining new or repurposed compounds for their potential as psychiatric medications. This mechanism will also investigate whether the compound engaged specific targets in the brain by altering brain signaling or a specific neurotransmitter. Importantly, no one group could possibly investigate each of these areas and so academia and industry must come together. These topics were covered in The International Society for CNS Clinical Trials and Methodology (ISCTM) Autumn 2013 meeting on the topic of translational and early development strategies and tools led by Drs. Potter and Feltner. This report comprises a review of those proceedings with a concluding summary to advance future clinical trials.
The Importance of Translational Pharmacology
Translational pharmacology is essential to ensure that the drug candidate is capable of testing the drug efficacy and hypothesis.[2] The goal, thereafter, is that the early preclinical pharmacological data will predict clinical efficacy. Numerous important aspects to measuring translational pharmacology should be considered, including plasma or cerebrospinal fluid levels, ex-vivo binding, micro-positron emission tomography (PET), and dose response curves. Table 1 provides a fictitious example of such considerations for a dopamine D1 receptor agonist. Ultimately, it is important for researchers to examine the pharmacology translation alongside the efficacy translation.
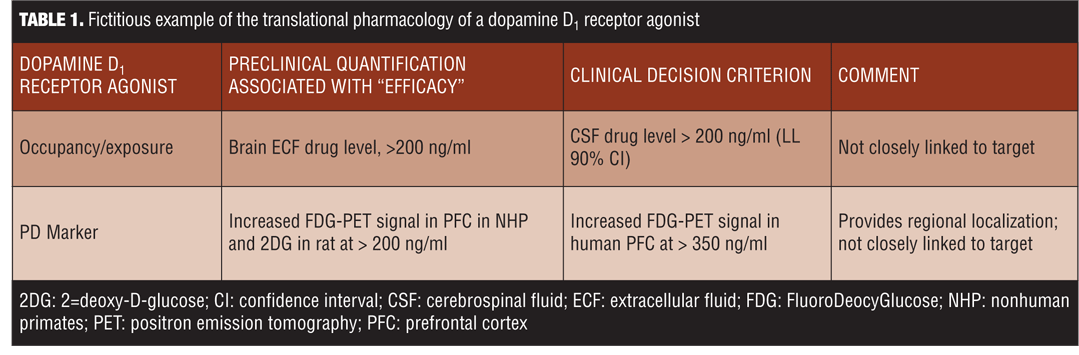
While conducting translational pharmacological research, there are numerous caveats and complexities that must be taken into account. For example, preclinical data on exposure and occupancy may be inconsistent so there would be less certainty in the translation to human data. Inverted u-shape dose response curves also exist, in both animals and humans, that require careful reviews of the data. Another complexity in translational pharmacology is off-target effects impacting biomarker and pharmacodynamic responses. Another important and often overlooked complication is whether there are differences between chronic or acute effects, and whether the drug would be used as an adjunctive treatment in already approved medications. Thus, drug interactions should be assessed when translating pharmacological effects. One other issue that often adds complexity is that the occupancy/exposure required in an animal model may differ from that required in human diseases. In fact, differences may also be seen between healthy humans and those suffering from disease. Ultimately, quantitative translation attempts to set a minimum exposure level to test the hypothesis and hence search for consistency in biomarker data, especially in novel targets. If this is not possible and current methodologies are the only path forward, then novel target development or rejection might be limited.
Go/No-Go Decision-making After Rigorous Early Phase Drug Evaluation
Such translational pharmacology can be pivotal in drug development. For instance, determining whether similar occupancy levels occur between preclinical and clinical testing can dictate the decision to move forward with or reject a target compound (go/no-go decisions).
One example of a go/no-go decision focuses on an early phase study in healthy subjects with a norepinephrine transport inhibitor that was to be developed as a new pain drug. The compound (A) was developed from a snake venom and was a small (1400 Da) stable 13-amino acid peptide that could not pass the blood brain barrier (BBB) and therefore had to be administered intrathecally. Because of its very high potency, it was being developed for severe chronic pain and potentially for postoperative pain. In initial Phase I studies, 20 healthy individuals were administered a single dose of compound A intravenously. The efficacy of compound A was examined in response to an earlobe electric stimulation test. Unfortunately, no pain-reducing efficacy of the compound was observed. In another early phase study examining its effects on patients with cancer, doses up to 40mg were given, which appeared to be beneficial. However the study was an uncontrolled, open-label design so definitive conclusions about the analgesic effects could not be made. More importantly, at the highest dose, there were two serious adverse events: an aseptic meningitis and a generalized epileptic seizure. Particularly, the potential epileptogenicity of the compound led to the FDA putting the development of the compound on halt when the company tried to get a study approved in 200 bunionectomy patients, which by then had already begun in Eastern Europe. Subsequently, the company decided to perform a safety study, aimed solely at the potential epileptogenicity by examining electro-encephalographic effects of the compound when administered to healthy subjects. By then, four studies in humans had been performed and still very little was known about the potential analgesic effects of the compound or about the pharmacokinetics in CSF. The final study investigated the effects of compound A at 0.5, 1, and 2.5mg (n=8 per dose) or placebo (n=8) administered intrathecally to healthy subjects . In this study, pharmacokinetics in the CSF were measured using a spinal catheter and CSF sampling over a period of 32 hours. Concurrently, the analgesic potential of the compound was determined, using a nociceptive test battery, which was administered multiple times after drug administration. The nociceptive test battery comprises two different paradigms of electrical pain,[3] pneumatic pain,[4] the cold pressor,[5] and thermal stimulation. Improved response to pain lasting up to 96 hours was observed at the highest dose (2.5mg) in two of the four nociceptive tests, providing evidence for the analgesic potential of the compound. However, at that dose level (2.5mg), CSF exposure as measured using 32-hour sampling turned out to be higher than expected and to exceed the safety limit that was defined in advance, based on the potential of the drug to cause epilepsy in dogs. Because of the findings in the study, it was decided to stop the development of this compound. This study is an example of a situation where failing earlier in the development may have saved time and cost.
In a second example, a partial GABA-A agonist was being developed for anxiety. Agonists at the alpha 1 receptor are linked to sedation of benzodiazepines, alpha 2 and alpha 3 subtype selective agonists are associated with muscle relaxation and anxiolytic effects, while the alpha 5 subunit is linked to cognitive impairments induced by GABA-A receptor agonists. In this study, an alpha 2/3 subtype selective GABA-A receptor agonist was tested in a central nervous system test battery lasting 20 to 25 minutes, including adaptive tracking, finger tapping, and 30-word learning. A dosage of 1.5mg of the new compound was found to be equipotent to 2.0mg lorazepam according to their influence on saccadic eye movements, a biomarker for the effects of benzodiazepines. At the equipotent dose of 1.5mg, however, the new compound had fewer sedative effects than 2.0mg of lorazepam. This could not have been predicted using PET, as the new compound had a 10-fold higher receptor occupancy compared with lorazepam. This is, therefore, a good example of the added value of pharmacodynamic testing when trying to benchmark a new compound compared to existing comparable drugs. Because of its apparent advantages in terms of lower sedation compared to lorazepam, this novel subtype selective GABA-A receptor agonist was taken forward in clinical development.
In conclusion, these examples demonstrate that measuring pharmacodynamic effects and trying to rationally assess the properties of a drug early in clinical drug development can greatly inform decisions for later stage development, which has implications on cost. Answering the questions with the highest uncertainty first in drug development may lead to lower overall development costs.[6]
Exposure response modeling in early development
Phase I testing can be conducted to be more informative during early development of compounds. Such an approach can facilitate quantitative bridging of nonclinical and clinical data. Experiments, therefore, need to be designed to be informative for this purpose. Such an approach can improve early and data-driven decision-making of the go/no-go status of a compound as well as aid in the interpretation of effects recorded in studies.
One example is to design Phase I experiments to better predict dose response and time course of a novel biomarker to a compound with an unprecedented mechanism of action. A road-map of the components used to predict human drug response is shown in Figure 1. As with any analysis methodology, there are assumptions with varying levels of uncertainty. In this particular case, the greatest uncertainty was the translatability of the dog PK/PD relationship to humans. Based upon the available data, it was predicted that 25mg would be a sufficient dose to demonstrate a drug effect. However, earlier studies suggested 25mg would not be tolerated and so the dose was dropped to 10mg. This lower dose was still predicted to show an effect, but at a much lower magnitude. Contrary to expectations, the compound lowered biomarker concentrations in humans. The reason for this effect was unclear, but could have stemmed from too low a dose, the unprecedented mechanism, misspecification of the model, or small sample size.

A second example comes from a proof of concept study in acute schizophrenia. The effects of Compound YYY with doses of 2, 5, and 15mg on PANSS total scores in patients with acute exacerbation of schizophrenia was compared with aripriprazole (15mg) as an active control. All treatments produced an improvement in PANSS score, but so did placebo. In fact, no separation of any treatments from placebo was observed using a linear trend test. Based upon a model-based meta-analysis of published acute schizophrenia data, the placebo effect was comparable with that of other publications, with simulations suggesting the results were plausible, although larger than reported by most studies. The effects of aripriprazole were also consistent with previous studies. Exposure-response analysis of Compound YYY data demonstrated that some components of the dose response, which would be critical for dose selection for the next study, were poorly characterized, thus identifying deficiencies in the learnings from the study.
The examination of the effects of Compound YYY resulted in several lessons learned. Small studies can result in variable point estimates and have diminished power, while linear trend tests may not be sufficiently powerful for certain dose-response curves. Dose response effects are not always linear so relying on a hypothesis testing approach with rigid assumptions in a small POC study may not be the most efficient use of resources for learning about a compound in early development. In conclusion, employing model-based analyses can identify and bridge knowledge gaps where they exist, and inform quantitative, data-driven decisions while extracting further information. Sufficient sample sizes and knowledge of the dose-response expectancies are required
Conducting the right proof-of-concept study
It is important to recognize that drugs could be successfully tested in numerous indications, e.g., an alpha-7 nicotinic acetylcholine receptor agonist could be useful for treating negative symptoms in schizophrenia and be pro-cognitive in patients with attention deficit hyperactivity disorder (ADHD) or Alzheimer’s disease. One case study comes from proof-of-concept studies Merck conducted for an H3 inverse agonist that targeted ADHD, was wake promoting in sleep apnea, and influenced cognition in Alzheimer’s disease and schizophrenia.
Numerous issues should be addressed to direct the development strategy, including target validation, how to assess target engagement and pharmacodynamics (PD) efficacy, and medical need. Examining data implicating the target in the disease pathophysiology is important. The clinical effects of H1 antagonists include sedation and cognitive impairment, with weak evidence of weight gain, although some of these effects may be due to off-target anticholinergic effects. There is preclinical evidence, however, suggesting that H1 agonists promote wakefulness. H3 antagonists act on inhibitory and autoreceptors.[7] ABT-239 is an H3 antagonist that had positive effects on numerous preclinical models of behavior, with improvements in sensory gating, five-choice serial reaction-time task, y-maze alternation, and radial arm maze,[8,9] although not always beneficial.[10] The approach of Merck with MK-0249 was to replicate some of these findings and combine them with developing translatable biomarkers for target engagement and PD effects.
In a Phase I program, multiple doses up to 12mg of MK-0249 were tolerated, with a half-life of 14 hours. Some insomnia effects were observed at higher doses, while PET studies showed 80- to 90-percent occupancy at 10mg. High occupancy was targeted but it is unclear what the optimal occupancy might be for the indications evaluated in Phase II. Electroencephalography studies in animals and humans demonstrated dose-response effects at higher frequencies, including gamma power, suggesting that an H3 antagonist may improve attention and other aspects of cognition. Another Phase I study examined the efficacy of MK-0249 on excessive daytime somnolence comparing its effects with modafinil (an approved treatment). While MK-0249 was shown to be superior to placebo, it was not as effective as modafinil. And another Phase I study examined whether MK-0249 would reverse scopolamine-induced deficits in healthy subjects using the Cog-State Battery. Scopolamine impaired reaction-time, choice reaction-time, executive functioning, as well as episodic and short-term memory. MK-0249 improved some scopolamine-induced deficits and had an additive effect with the Alzheimer’s disease treatment donepezil for some cognitive domains, although notably not episodic memory.[11] These positive results from phase I indicated clinically relevant target engagement and pharmacodynamics efficacy relevant and suggested that MK-0249 could improve symptoms in disorders of wakefulness and cognition.
Unfortunately, in several Phase II trials in a variety of indications, MK-0249 did not demonstrate sufficient efficacy to warrant further development. In a study of subjects with daytime somnolence due to sleep apnea, MK-0249 did not provide beneficial effects compared to placebo.12 In contrast, the active modafinil was superior to placebo, demonstrating the conduct of the trial was sufficient to detect efficacy. In a second Phase II trial of adult ADHD, MK-0249 improved exploratory measures of attention and executive function, but not the primary outcome measure, the ADHD Investigator Symptom Rating Scale (J. Herring et al., unpublished). In a third Phase II trial, MK-0249 treatment did not improve cognition in patients with schizophrenia on a summary score of cognitive tests or individual cognitive domains. Some subjects in this trial were receiving antipsychotic medications, which had H1 antagonist effects, which could have reduced the trial’s power to detect a therapeutic effect.[13] Finally, MK-0249 was tested in patients with mild to moderate Alzheimer’s disease. No effect was observed on any cognitive scores using a computerized test batter. A trend for a positive effect on a secondary measure, the ADAS-Cog, was seen at two but not four weeks.[14] Despite substantial evidence from preclinical studies as well as robust evidence of target engagement and pharmacodynamics efficacy in Phase I, no clinically significant therapeutic effects were seen in four Phase II trials in different disorders.
It is clear that multiple POC studies using one compound and overlapping biomarker sets was an efficient use of resources providing information on multiple indications. The current studies also demonstrate, however, that evidence of target validation was insufficient. In the future, successful drug development in neuropsychiatry may depend on stronger support for a drug target. It seems likely that animal models are poorly predictive of efficacy in humans, although efficacy in animal models may be necessary but not sufficient. In terms of biomarkers, CNS drug development programs should ideally include both measures of target engagement as well as pharmacodynamic efficacy. Future studies should focus on targets with substantial evidence, particularly genetic evidence, that the target is implicated in the critical disease process. In Alzheimer’s disease, for example, the beta secretase enzyme is a good target with CSF A-beta serving as a potential pharmacodynamics biomarker.
Optimizing Phase II studies to set the stage for success in Phase III trials
Optimizing each stage in the drug discovery process would not necessarily optimize drug development equally, but optimizing Phase II likely has the greatest impact. Minimizing the placebo response is also important for trial optimization. Three pillars have been identified as important for enhancing treatment development: 1) exposure at the target, 2) pharmacological activity commensurate with target exposure, and 3) binding to the target.[2] When these three pillars are all met, the likelihood of clinical development success is enhanced. The framework for choosing a plan have been previously explored and known as the axes of development. Efficient Phase IIa POC studies followed by a Phase IIb study can focus on the quick termination of a target. With high optimization and effect sizes, registration of the compound could be fast-tracked. This strategy also provides a dose-response curve of the treatment being developed.
Using positive controls can be beneficial but the conditions for such a control need to be understood. The power for the positive control must be high so that a failure of the treatment to separate from placebo is not by chance. Positive controls can reduce the sensitivity of the analyses with changes from control perhaps due to less effective placebos or unequal distribution to groups.
When conducting studies with multiple dose levels, interim analyses can enable dosing decisions throughout the study (e.g., identifying the “best dose” or eliminating doses altogether). Moreover, model-based analyses can be more efficient than straight-forward pairwise comparisons. The power gained from these small dose-finding Phase II studies can be used to determine the number of subjects required for Phase III success of the compound. This sample-size issue is important as protection to avoid false negative findings. Hence, there are multiple aspects to Phase II trials that require optimization and constant attention during drug development.
Optimizing Phase II trials remains critically important. An overall development plan should be in place and the portfolio of the compound should be considered. Compounds that meet the three-pillars criterion should be favored over those that do not. Ultimately, enhancing this design – and an understanding of effects from earlier phases – will likely result in targeting the most likely agents to be successfully developed.
Conclusion
In summary, there are a wide range of strategies aimed at optimizing the drug development process. Specifically, the translation of compounds from Phase I to Phase III clinical trials was emphasized here. The translational pharmacology, including PK/PD data should be quantitatively characterized across species. Stringent a priori decision-making criteria should be identified to aid go/no-go decision-making for a compound. Furthermore, the tests used when translating preclinical to Phase I through III studies should be as consistent as possible, enhancing the likelihood that the same mechanism is being assessed when taking the treatment into human testing. Another critical point is to employ model-based analyses to understand dose-response and effect sizes of particular doses. Utilizing biomarker data to examine efficacy of a compound across disease states may also be an efficient strategy and use of resources. Finally, while using a positive control is vital in many studies, the strength of this control should be carefully weighed against possible costs. In summary, while treatment development for serious mental illness sufferers has been limited, many lessons have been learned and future studies will be more informative for these complex diseases.
References
1. Morris SE, Cuthbert BN. Research Domain Criteria: cognitive systems, neural circuits, and dimensions of behavior. Dialogues Clin Neurosci. 2012;14:29–37.
2. Morgan P, Van Der Graaf PH, Arrowsmith J, et al. Can the flow of medicines be improved? Fundamental pharmacokinetic and pharmacological principles toward improving Phase II survival. Drug Discov Today. 2012;17:419–424.
3. Olofsen E, Romberg R, Bijl H, et al. Alfentanil and placebo analgesia: no sex differences detected in models of experimental pain. Anesthesiology. 2005;103:130–139.
4. Polianskis R, Graven-Nielsen T, Arendt-Nielsen L. Computer-controlled pneumatic pressure algometry–a new technique for quantitative sensory testing. Eur J Pain. 2001;5:267–277.
5. Jones SF, McQuay HJ, Moore RA, Hand CW. Morphine and ibuprofen compared using the cold pressor test. Pain. 1988;34:117–122.
6. Cohen AF. Developing drug prototypes: pharmacology replaces safety and tolerability? Nat Rev Drug Discov. 2010;9:856–865.
7. Esbenshade TA, Fox GB, Cowart MD. Histamine H3 receptor antagonists: preclinical promise for treating obesity and cognitive disorders. Mol Interv. 2006;6:77–88, 59.
8. Kruk M, Miszkiel J, McCreary AC, Przegalinski E, Filip M, Biala G. Effects of the histamine H(3) receptor antagonist ABT-239 on cognition and nicotine-induced memory enhancement in mice. Pharmacol Rep. 2012;64:1316–1325.
9. Brown JW, Whitehead CA, Basso AM, Rueter LE, Zhang M. Preclinical evaluation of non-imidazole histamine H3 receptor antagonists in comparison to atypical antipsychotics for the treatment of cognitive deficits associated with schizophrenia. Int J Neuropsychopharmacol. 2013;16:889–904.
10. Burban A, Sadakhom C, Dumoulin D, et al. Modulation of prepulse inhibition and stereotypies in rodents: no evidence for antipsychotic-like properties of histamine H3-receptor inverse agonists. Psychopharmacology (Berl). 2010;210:591–604.
11. Cho W, Maruff P, Connell J, et al. Additive effects of a cholinesterase inhibitor and a histamine inverse agonist on scopolamine deficits in humans. Psychopharmacology (Berl). 2011;218:513–524.
12. Herring WJ, Liu K, Hutzelmann J, et al. Alertness and psychomotor performance effects of the histamine-3 inverse agonist MK-0249 in obstructive sleep apnea patients on continuous positive airway pressure therapy with excessive daytime sleepiness: a randomized adaptive crossover study. Sleep Med. 2013;14:955–963.
13. Egan MF, Zhao X, Gottwald R, et al. Randomized crossover study of the histamine H3 inverse agonist MK-0249 for the treatment of cognitive impairment in patients with schizophrenia. Schizophr Res. 2013;146:224–230.
14. Egan M, Yaari R, Liu L, et al. Pilot randomized controlled study of a histamine receptor inverse agonist in the symptomatic treatment of AD. Curr Alzheimer Res. 2012;9:481–490.