
 by Nazanin Arjomand Fard, MSci; Gholamreza Azizi, PhD; and Abbas Mirshafiey, PhD
by Nazanin Arjomand Fard, MSci; Gholamreza Azizi, PhD; and Abbas Mirshafiey, PhD
Dr. Fard is with Kish International Campus, University of Tehran, Tehran, Iran; Dr. Azizi is with Imam Hassan Mojtaba Hospital, Alborz University of Medical Sciences, Karaj, Iran and Research Center for Immunodeficiencies, Children’s Medical Center, Tehran University of Medical Sciences, Tehran, Iran; and Dr. Mirshafiey is with the Department of Immunology, School of Public Health, Tehran University of Medical Sciences, Tehran, Iran.
Innov Clin Neurosci. 2016;13(7–8):30–36.
Funding: No funding was received for the preparation of this article.
Financial disclosures: The authors have no conflicts of interest relevant to the content of this article.
Key words: T helper 22, interleukin-22, multiple sclerosis
Abstract: Multiple sclerosis is a complex disease with many different immune cells involved in its pathogenesis. Newly identified T helper cell 22 (Th22) is a subset of CD4+ T cells with specific properties apart from other known CD4+ T cell subsets with distinguished function and gene expression. Th22 cells are characterized by production of a distinct profile of effector cytokines, including interleukin (IL)-22, IL-13, and tumor necrosis factor-a (TNF-alpha). The frequency of Th22 and related cytokine IL-22 are increased in various autoimmune diseases. Recently, several studies have reported the changes in frequency and function of Th22 in multiple sclerosis. This review discusses the role of Th22 and its cytokine IL-22 in the immunopathogenesis of multiple sclerosis disease.
Introduction
Multiple sclerosis (MS) is a chronic inflammatory autoimmune disease of the central nervous system (CNS) with unknown etiology and heterogeneous clinical symptoms and course.[1] Demyelination, where the myelin sheath or the oligodendrocyte cell body is destroyed by the inflammatory process, is the hallmark of MS. Myelin plays a critical role in insulation of nerve fibers and regulates the speed of neurotransmission, and myelin destruction is seen in numerous neurological disorders, including MS.[2,3] MS is a complex disease with many different immune cells involved in its pathogenesis. The cellular and homoral arms of the immune system have critical roles in the pathogenesis of MS, especially on adaptive immune system. Recent studies have suggested that the innate immune system also plays an important role both in the initiation and progression of MS by influencing the effector function of T and B cells.[4–6] In particular, T cells are the most recognized cell type, and inflammatory lesions in the CNS of MS patients contain both CD4+ and CD8+ T cells.[7] In addition, studies have shown that MS patients have a significant loss of effector function in the regulatory CD4+CD25+ subset of T cells (Treg).[8] Myelin-specific T cell clones, including T helper 17 (Th17) cells and their pro-inflammatory cytokines IL-17 and IL-6, along with Th1 and its cytokines IL-2, IFN-gamma, and TNF-alpha have been found in MS lesions.[9,10] In addition, B cells and plasma cells can also be found in MS lesions.[11] Th22 cells as a new CD4+ T cell subset are mainly found in tissues. These cells and their cytokine (IL-22) levels are increased in autoimmune diseases such as rheumatoid arthritis, psoriasis, and MS. The main targets of the autoimmune reactions are thought to be myelin basic protein (MBP), proteolipid protein (PLP), and myelin oligodendrocyte glycoprotein (MOG).[12] In this review, we will focus on the role of Th22 and its pro-inflammatory cytokine IL-22 in the pathogenesis of MS.
T helper 22
Th22 cells are a subset of CD4+ effector T cells that link to the innate immune response and inflammation in autoimmune disorder; however, the accurate role of this subset in autoimmune disorder is not fully understood. Th22 cells secrete IL-22, IL-13, and TNF-alpha. They also express other factors such as fibroblast growth factor isoforms, which contribute to tissue remodeling, and chemokine receptors CCR4, CCR6, and CCR10. In addition, they do not express IL-17, IL-23R, CCL20, IFN-g (Th1 marker) or IL-4 (Th2 marker). Furthermore, Th22 cell subset is distinguished from Th17 cells by low or no CD161 (Th17 markers) expression and a high level of poly functionality.[13,14] Duhen et al[15] reported that Th22 has low or undetectable expression of the Th17 and Th1 transcription factors RORgt, GATA-3, and T-bet. Thus, these features distinguish Th22 cells from the Th17, Th2, and Th1 subtypes. In other words, Th22 cells are a novel Th cell lineage.[13–15] Activated naive CD4+ T cells can differentiate into Th22 cells in response to TNF-alpha and IL-6. The expansion of Th22 cells seems to be regulated by a transcription factor named aryl hydrocarbon receptor (AHR) (Table 1).[16,17] The differentiation process could be also inhibited by the addition of increased concentrations of TGF-beta.[18]
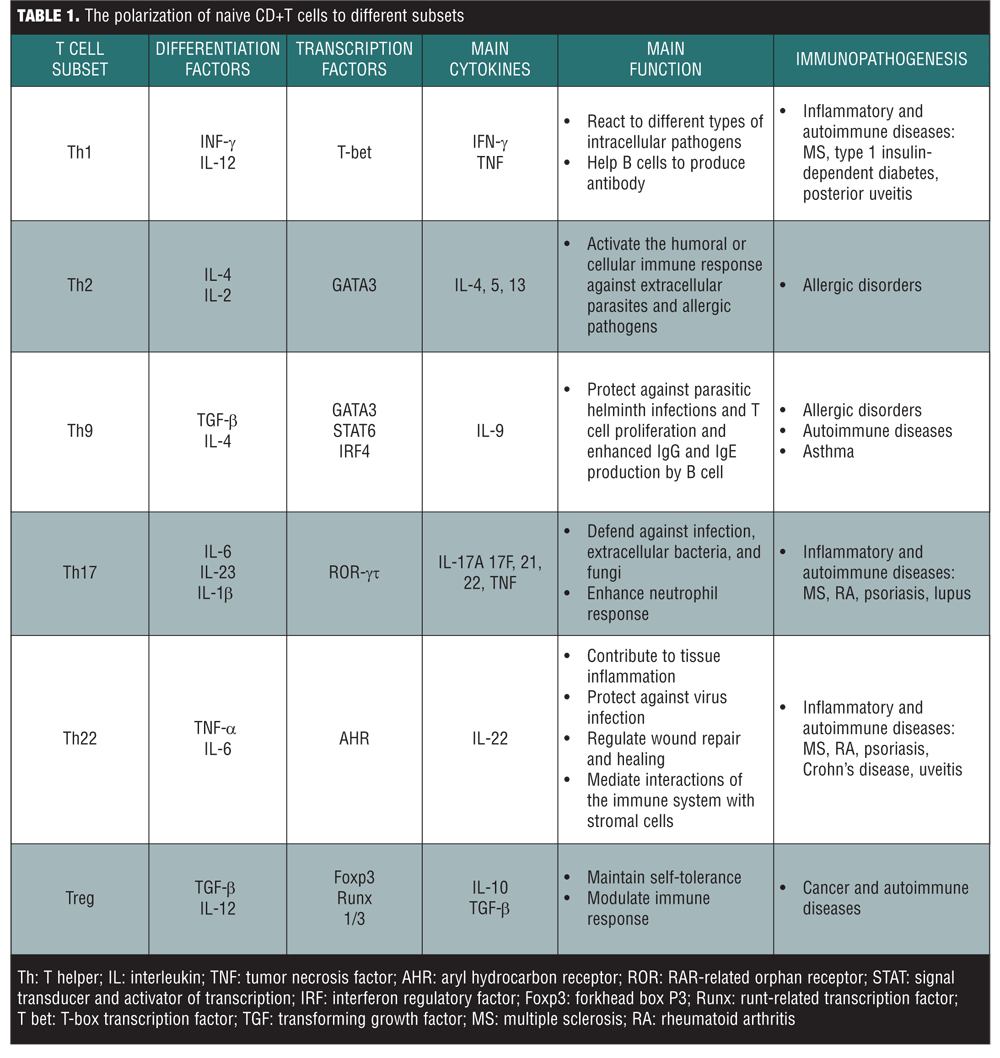
The notch family of cell membrane receptors is a vital modulator of T cell-mediated immune responses. In the last decade, evidence has shown that notch cells via their interactions with the canonical nuclear binding protein CSL/RBP-J have important roles as transcriptional regulators in T cell activation.[19] It was demonstrated that via induction of endogenous AHR stimulators, notch signaling drives the production of IL-22. In addition, it is suggested that induction of CD4+ T cells by notch result in elevation of the IL-22, even in the absence of STAT3.[20] Also, down-regulation of either the transcription factors AHR or the RORC by RNA-mediated interference affect IL-22 production, whereas IL-17 secretion is affected just by down-regulation of RORC through RNA-mediated interference. In other word, AHR agonists reduce the expression of the Th17 master transcription factor RORC without affecting T-bet, GATA-3, or Foxp3. AHR ligation not only decreases the Th17 cell number, but also primes naive CD4+ T cells to produce IL-22 without IL-17 and IFN-gamma.[21] Finally, there is clear evidence that AHR activation participates in shaping human CD4+ T-cell polarization, favoring the emergence of a Th22 distinct subset. Similar to Th17 cells, Th22 cells and its main cytokine IL-22 are involved in the pathogenesis of inflammatory and autoimmune diseases.[22]
Interleukin 22
IL-22 is part of the IL-10 cytokine family, which includes IL-10, IL-19, IL-20, IL-22, IL-24, and IL-26 and the more distantly related IL-28A, IL-28B, and IL-29.[23,24] Like other IL-10 family members, IL-22 has also an a-helical secondary structure.[25] In contrast to IL-10, IL-22 does not prevent the secretion of pro-inflammatory cytokines by monocytes in response to LPS and also nor does the function of IL-10 on monocytes, but it has a modest inhibitory effects on IL-4 secretion from Th2 cells.[26] IL-22 was originally thought to be a Th1-associated cytokine; now it is recognized that IL-22 is highly expressed by Th17 and Th22 cells. Several innate immune cells, such as mast cells, CD11c+ DCs, gd T cells, NKT cells, natural killer22 (NK22) cells, lymphoid tissue inducer (LTi) and LTi-like cells, are also enabled to produce IL-22 (Figure 1). It can be also produced by CD8+ T cells.[23,27,28]
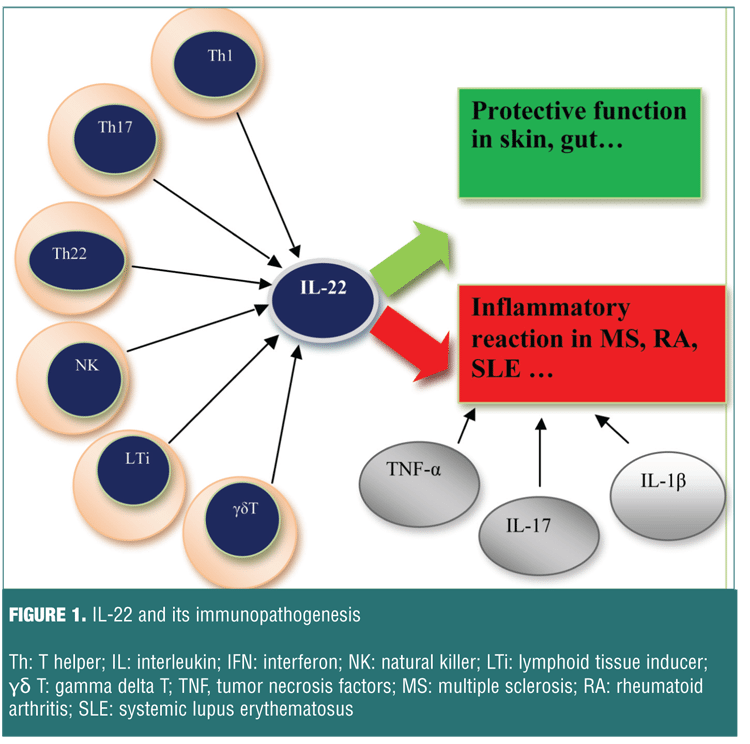
IL-22 not only raises pro-inflammatory innate defense mechanisms in epithelial cells, but also provides vital protection to tissues from damage produced by inflammation and infection.[29] As it mentioned before, the most important IL-22 producing cells are the Th-22 cell. IL-22 exerts its effect on epithelial and stromal cells in the skin, gut, and lung.[30] In the other words, in contrast to its inflammatory role in autoimmune diseases, IL-22 also plays a protective role in the prevention of inflammation in other diseases.[31,32] Thus, there is a paradox about the role of IL-22 in the immune system. Recent findings have revealed that IL-22 is differentially expressed in many autoimmune diseases.[33,34] In fact, depending on the target tissue, IL-22 is beneficial to the host in many infectious and inflammatory disorders, and it could be pathogenic due to its inherent pro-inflammatory properties, which are further enhanced when IL-22 is secreted with other pro-inflammatory cytokines, in particular IL-17.[35]
IL-22 binds to a heterodimer class II cytokine receptor complex composed of the IL-10R2 and the IL-22R1. IL-22R1 is expressed in a variety of non-immune tissues, such as lung, kidney, skin, and pancreas, while the expression of IL-10R2 is widely present in immune cells, such as T, B, and NK cells.[27] There is also a soluble IL-22R called IL-22-binding protein (IL-22BP). This protein acts as a natural, soluble antagonist of IL-22. The complex of IL-22 and IL-22R induces a cascade of downstream signaling pathways.[36] IL-22 signal transduction pathways show that IL-22 binding to its receptor leads to activation of Tyk2 and JAK1 but not JAK2, as well as phosphorylation of STAT1, STAT3, and STAT5. A common pathways shared by the IL-10 cytokine family members is STAT3-mediated signaling; IL-22 signaling displays several distinct properties. Lejeune et al[37] have found that IL-22 triggers the three major MAPK pathways (ERKs, JNKs and p38/SAPKs) via phosphorylation of MEK1/2, ERK1/2, p90RSK, JNK, and p38 kinase. In the other words, IL-22 strongly activates the ERK1/2 pathway via stimulation of STAT3 phosphorylation on both serine and tyrosine residues.[37,38] Some factors, such as c-Maf act specifically on IL-22 and induce the expression of other cytokines while common factors such as STAT3 and RORgt drive the expression of both IL-22 and IL-17 cytokines.[39] Rutz et al[40] have demonstrated that transcription factor c-Maf is induced by TGF-beta as a downstream repressor of IL-22. They have reported that binding c-Maf to the IL-22 promoter in Th17 cells was both essential and sufficient for the TGF-beta-dependent suppression of IL-22 secretion.[40] Therefore, manipulation of the cellular source of IL-22 and/or its signaling pathways may provide a therapeutic target for autoimmune diseases such as MS.
T Helper 22, Interleukin-22, and MS
Th22 cells were found to be dedicated to the autoantigen myelin basic protein, which is important in the process of myelination of nerves in the nervous system. IL-22 is also involved in the pathogenesis of experimental autoimmune encephalomyelitis (EAE), a murine model of human MS. The role of IL-22 in MS has not yet been elucidated but it has been done investigations in this connection, in recent years.[41,42]
The EAE as an MS model is often used to study neuroinflammatory disease mechanisms. The recent advances in whole-genome screening tools via this model have enabled discovery of several MS risk genes, the majority of which are known as immune-related factors. A single nucleotide polymorphism of IL-22 receptor (IL-22RA2) is a risk gene in EAE and in patients with MS. Another single-nucleotide polymorphisms close to IL22RA2 and coding for the soluble IL-22-binding protein (IL-22BP) are also strongly associated with MS. IL-22BP is known to antagonize IL-22 signaling, which is a primarily proinflammatory cytokine. Beyeen et al[43] in 2010 detected this risk gene through a combined approach that included immunological and genetic investigation in an animal model and large-scale association studies of MS patients, which established IL-22RA2 as an MS risk gene. In a similar study in 2014, Laaksonen et al[44] demonstrated that lack of IL-22BP leads to a higher accessibility of IL-22, which in CNS inflammation, surprisingly acts in a protective fashion. In other words, IL-22 and its receptors have been implicated in chronic inflammation, suggesting that IL-22RA2 regulates a central immune pathway.[44,45]
In recent studies, the roles of this IL-22 and Th22 have been investigated in MS patients. There is evidence that serum IL-22 levels were increased and Th22 cells were activated in patients with MS during a relapse phase. Th22 cells are correlated with Th17 cells, suggesting that Th22 cells and Th17 cells may play a synergistic role in MS progression.[45] In 2014 Rolla et al[41] showed that similar to Th17, the number of Th22 cells was increased in the peripheral blood and the CSF of patients with relapsing-remitting MS (RRMS), especially during the active phases of the disease.[41] It is likely that the expansion of Th22 cells contribute to breach the blood brain barrier, allowing increased T cell infiltration that triggers the MS disease.[46] IFN-gamma therapy is the most commonly used treatment for RRMS patients.[47–49] Interestingly, it is revealed that one of the factors that could critically influence resistance to IFN-gamma therapy is the increase of Th22 cells, which occurs before the active phase of disease. Rolla et al[41] showed that Th22 cells display lower levels of IFNgR1 and are insensitive to IFN-gamma therapy; therefore, these MS patients are resistant to this therapy. These cells also express high levels of CCR6 and T-bet, which indicates that Th22 self-reactive cells could have CNS-homing properties and be pathogenic in patients with active RRMS. Thus in MS pathogenesis, these cells are thought to initiate an autoimmune response directed against components of CNS myelin.[41,46] In 2013, Xu et al[45] demonstrated that the proportion of Th22 cells and Th17 cells in MS patients was higher than in control samples. Serum IL-22 levels are also increased in MS patients, indicating that the disease duration positively correlates with Th22 cells in MS. The proportion of Th22 cells and Th17 cells are also both elevated in the PBMCs of patients with MS.[45] In a similar study in 2011, Almolda et al[50] observed a marked decrease of Th1 and a notable increase in T-reg and Th17 cells. Also, IL-22 concentrations were seen to sharply decrease during the recovery phase of MS?along the EAE course. Moreover, the results of the study indicate a specific cytokine expression profile characterized by no changes of IL-10 and IL-17 levels, decrease in IL-21 on the peak, and high IL-22 levels during the induction, whereas IL-22 levels significantly decrease during recovery phase.[50] Studies on MS along the EAE course revealed for the first time that the proportion of Th22 cells, Th17 cells, and serum IL-22 levels are increased in patients with MS.[51]
Recently, Perriard et al[52] demonstrated the same result, finding that the serum levels of IL-22 are significantly higher in patients with relapsing MS than in healthy controls. They also showed that astrocytes in the human brain express the both subunits of IL-22 receptor, and there is a colocalization of IL-22 with these cells, which leads to pro-survival properties on primary human astrocytes.[52] Astrocytes have been recognized as being an important component of the pathogenesis of MS disease.[53]
Studies that have shown that some drugs, such as simvastatin, can influence the secretion of IL-22 cytokine. Simvastatin, one of the most hydrophobic statins, is presented as an inhibitory factor for secreting IL-22 and IL-17A and other inflammatory cytokines in patients with RRMS. These studies reported that simvastatin can inhibit Th17 cell differentiation and IL-17A, IL-17F, IL-21, and IL-22 secretion in differentiated naive CD4(+) T cells in patients with RRMS due to its ability to penetrate the CNS.[45,54] It is interesting to note that there was a marked decrease in the levels of IL-22 in the initiation of the recovery phase. This prominent result opens a new and interesting way for evaluating IL-22 as a putative key factor involved in the evolution of EAE. Therefore, the increased Th22 cells and serum IL-22 may play an important role immunopathogenesis of MS, similar to Th17 cells. One feasibility is that this cytokine may drive the inflammatory events occurring during the peak and inductive phases of MS, and accordingly, its decrease in production may stop the inflammation and initiate recovery. In other words, IL-22 may play an important role in the pathogenesis of both MS and EAE.[50]
Conclusion
Recent evidence shows changes in frequency and function of Th22 and its cytokine IL-22 in patients with MS, which suggests a potential relationship between Th22 cells, IL-22 levels, and the development and disease course of MS and its response to treatment. How Th22 cells and IL-22 levels affect the disease course of MS requires further elucidation. Additional studies on the immunopathogenesis of MS, specifically the roles Th22 cells and IL-22 cytokines may play in disease progression, are warranted and may lead to the development of new therapeutic strategies for treating neuro-inflammatory diseases, such as MS.
References
1. Weiner HL. Multiple sclerosis is an inflammatory T-cell–mediated autoimmune disease. Arch Neurol. 2004;61(10):1613–1615.
2. O’Brien K, Gran B, Rostami A. T-cell based immunotherapy in experimental autoimmune encephalomyelitis and multiple sclerosis. Immunotherapy. 2010;2(1):99–115.
3. R&D Systems website. Multiple sclerosis: immune cell access to the central nervous system. 2006. https://www.rndsystems.com/resources/articles/multiple-sclerosis-immune-cell-access-central-nervous-system. Accessed June 15, 2016.
4. Gandhi R, Laroni A, Weiner HL. Role of the innate immune system in the pathogenesis of multiple sclerosis. J Neuroimmunol. 2010;221(1):7–14.
5. Constantinescu CS, Farooqi N, O’Brien K, Gran B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br J Pharmacol. 2011 Oct;164(4):1079–1106.
6. Weiner HL. The challenge of multiple sclerosis: how do we cure a chronic heterogeneous disease? Ann Neurol. 2009;65(3):239–248.
7. Traugott U, Lebon P. Multiple sclerosis: involvement of interferons in lesion pathogenesis. Ann Neurol. 1988;24(2):243–251.
8. Viglietta V, Baecher-Allan C, Weiner HL, Hafler DA. Loss of functional suppression by CD4+ CD25+ regulatory T cells in patients with multiple sclerosis. J Exp Med. 2004;199(7):971–979.
9. Lucchinetti C, Brück W, Parisi J, et al. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol. 2000;47(6):707–717.
10. Storch MK, Piddlesden S, Haltia M, et al. Multiple sclerosis: in situ evidence for antibody- and complement-mediated demyelination. Ann Neurol. 1998;43(4):465–471.
11. Genain CP, Cannella B, Hauser SL, Raine CS. Identification of autoantibodies associated with myelin damage in multiple sclerosis. Nat Med. 1999;5(2):170–175.
12. Grigoriadis N, Hadjigeorgiou GM. Virus-mediated autoimmunity in multiple sclerosis. J Autoimmune Dis. 2006;3(1).
13. Larsen M, Arnaud L, Hié M, et al. Multiparameter grouping delineates heterogeneous populations of human IL17 and/or IL22 Tcell producers that share antigen specificities with other Tcell subsets. Eur J Immunol. 2011;41(9):2596–2605.
14. Trifari S, Kaplan CD, Tran EH, et al. Identification of a human helper T cell population that has abundant production of interleukin 22 and is distinct from TH-17, TH1 and TH2 cells. Nat Immunol. 2009;10(8):864–871.
15. Duhen T, Geiger R, Jarrossay D, et al. Production of interleukin 22 but not interleukin 17 by a subset of human skin-homing memory T cells. Nat Immunol. 2009;10(8):857–863.
16. Baba N, Rubio M, Kenins L, et al. The aryl hydrocarbon receptor (AhR) ligand VAF347 selectively acts on monocytes and naïve CD4< sup>+ Th cells to promote the development of IL-22-secreting Th cells. Hum Immunol. 2012;73(8):795–800.
17. Eyerich S, Eyerich K, Pennino D, et al. Th22 cells represent a distinct human T cell subset involved in epidermal immunity and remodeling. J Clin Invest. 2009;119(12):3573–3585.
18. Cheng R. [Expression level of Th22 cells and its cytokines in patients with acute lymphoblastic leukemia and its significance]. Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2013;21(4):857–860.
19. Minter LM, Osborne BA. Canonical and non-canonical Notch signaling in CD4(+) T cells. Curr Top Microbiol Immunol. 2012;360:99–114.
20. Alam MS, Maekawa Y, Kitamura A, et al. Notch signaling drives IL-22 secretion in CD4+ T cells by stimulating the aryl hydrocarbon receptor. Proc Natl Acad Sci U S A. 2010;107(13):5943–5948.
21. Ramirez JM, Brembilla NC, Sorg O, et al. Activation of the aryl hydrocarbon receptor reveals distinct requirements for IL-22 and IL-17 production by human T helper cells. Eur J Immunol. 2010;40(9):2450–2459.
22. Trifari S, Kaplan CD, Tran EH, et al. Identification of a human helper T cell population that has abundant production of interleukin 22 and is distinct from T(H)-17, T(H)1 and T(H)2 cells. Nat Immunol. 2009;10(8):864–871.
23. Sabat R. IL-10 family of cytokines. Cytokine Growth Factor Rev. 2010;21(5):315–324.
24. Pestka S, Krause CD, Sarkar D, et al. Interleukin-10 and related cytokines and receptors. Annu Rev Immunol. 2004;22:929–979.
25. Wolk K, Sabat R. Interleukin-22: a novel T-and NK-cell derived cytokine that regulates the biology of tissue cells. Cytokine Growth Factor Rev. 2006;17(5):367–380.
26. Zhou L, Chong MM, Littman DR. Plasticity of CD4< sup>+ T cell lineage differentiation. Immunity. 2009;30(5):646–655.
27. Cella M, Fuchs A, Vermi W, et al. A human natural killer cell subset provides an innate source of IL-22 for mucosal immunity. Nature. 2009;457(7230):722–725.
28. Nograles KE, Zaba LC, Shemer A, et al. IL-22–producing “T22” T cells account for upregulated IL-22 in atopic dermatitis despite reduced IL-17–producing T H 17 T cells. J Allergy Clin Immunol. 2009;123(6):1244–1252. e2.
29. Rutz S, Noubade R, Eidenschenk C, et al. Transcription factor c-Maf mediates the TGF-beta-dependent suppression of IL-22 production in T(H)17 cells. Nat Immunol. 2011;12(12):1238–1245.
30. Duhen T, Geiger R, Jarrossay D, et al. Production of interleukin 22 but not interleukin 17 by a subset of human skin-homing memory T cells. Nat Immunol. 2009;10(8):857–863.
31. Zenewicz LA, Yancopoulos GD, Valenzuela DM, et al. Interleukin-22 but not interleukin-17 provides protection to hepatocytes during acute liver inflammation. Immunity. 2007;27(4):647–659.
32. Radaeva S, Sun R, Pan HN, et al. Interleukin 22 (IL?22) plays a protective role in T cell?mediated murine hepatitis: IL?22 is a survival factor for hepatocytes via STAT3 activation. Hepatology. 2004;39(5):1332–1342.
33. da Rocha LF Jr, Duarte ÂL, Dantas AT, et al. Increased serum interleukin 22 in patients with rheumatoid arthritis and correlation with disease activity. J Rheumatol. 2012;39(7):1320–1325.
34. Cheng F, Guo Z, Xu H, et al. Decreased plasma IL22 levels, but not increased IL17 and IL23 levels, correlate with disease activity in patients with systemic lupus erythematosus. Ann Rheumat Dis. 2009;68(4):604–606.
35. Rutz S, Eidenschenk C, Ouyang W. IL-22, not simply a Th17 cytokine. Immunol Rev. 2013;252(1):116–132.
36. Sonnenberg GF, Fouser LA, Artis D. 1 functional biology of the IL-22-IL-22R pathway in regulating immunity and inflammation at barrier surfaces. Adv Immunol. 2010;107:1.
37. Lejeune D, Dumoutier L, Constantinescu S, et al. Interleukin-22 (IL-22) activates the JAK/STAT, ERK, JNK, and p38 MAP kinase pathways in a rat hepatoma cell line Pathways that are shared with and distinct from IL-10. J Biologic Chem.2002;277(37):33676–33682.
38. Dhillon AS, Hagan S, Rath O, Kolch W. MAP kinase signalling pathways in cancer. Oncogene. 2007;26(22):3279–3290.
39. Rutz S, Eidenschenk C, Ouyang W. IL22, not simply a Th17 cytokine. Immunological Reviews. 2013;252(1):116–132.
40. Rutz S, et al. Transcription factor c-Maf mediates the TGF-b-dependent suppression of IL-22 production in T(H)17 cells. Nat Immunol. 2011;12(12):1238–1245.
41. Rolla S, Bardina V, De Mercanti S, et al. Th22 cells are expanded in multiple sclerosis and are resistant to IFN-b. J Leukoc Biol. 2014;96(6):1155–1164.
42. Grigorian A, Araujo L, Naidu NN, et al. N-acetylglucosamine inhibits T-helper 1 (Th1)/T-helper 17 (Th17) cell responses and treats experimental autoimmune encephalomyelitis. J Biologic Chem. 2011;286(46):40133–40141.
43. Beyeen AD, Adzemovic MZ, Ockinger J, et al. IL-22RA2 associates with multiple sclerosis and macrophage effector mechanisms in experimental neuroinflammation. J Immunol. 2010;185(11):6883–6890.
44. Laaksonen H, Guerreiro-Cacais AO1, Adzemovic MZ, et al. The multiple sclerosis risk gene IL22RA2 contributes to a more severe murine autoimmune neuroinflammation. Genes Immun. 2014;15(7):457–465.
45. Xu W, Li R, Dai Y, et al. IL-22 secreting CD4+ T cells in the patients with neuromyelitis optica and multiple sclerosis. J Neuroimmunol. 2013;261(1):87–91.
46. De Mercanti S, Clerico M, Rolla S, et al. Role of Th22 expansion in multiple sclerosis disease activity. Presented at 29th Congress of the European Committee for treatment and Research in Multiple Sclerosis. Copenhagen, Denmark. 2–5 October 2013.
47. Hemmer B, Hartung HP. Toward the development of rational therapies in multiple sclerosis: what is on the horizon? Ann Neurol. 2007;62(4):314–326.
48. Compston A. The genetics of multiple sclerosis. Clin Chem Lab Med. 2000;38(2):133–135.
49. Fingerprinting G. Risk alleles for multiple sclerosis identified by a genomewide study. N Engl J Med. 2007;357:851–862.
50. Almolda B, Costa M, Montoya M, et al. Increase in Th17 and T-reg lymphocytes and decrease of IL22 correlate with the recovery phase of acute EAE in rat. PLoS One. 2011;6(11):e27473.
51. Maddur MS, Miossec P, Kaveri SV, Bayry J. Th17 cells: biology, pathogenesis of autoimmune and inflammatory diseases, and therapeutic strategies. Am J Pathol. 2012;181(1):8–18.
52. Perriard G, Mathias A1, Enz L, et al. Interleukin-22 is increased in multiple sclerosis patients and targets astrocytes. J Neuroinflammation. 2015;12(1):119.
53. Brosnan CF, Raine CS. The astrocyte in multiple sclerosis revisited. Glia. 2013;61(4):453–465.
54. Zhang X, Tao Y, Troiani L, Markovic-Plese S. Simvastatin inhibits IFN regulatory factor 4 expression and Th17 cell differentiation in CD4+ T cells derived from patients with multiple sclerosis. J Immunol. 2011;187(6):3431–3437.