
 by Atmaram Yarlagadda, MD; Ganesh Acharya, MD, PhD, FRCOG; Jayaprada Kasaraneni, MBBS; Christiane S. Hampe, PhD; and Anita H. Clayton, MD
by Atmaram Yarlagadda, MD; Ganesh Acharya, MD, PhD, FRCOG; Jayaprada Kasaraneni, MBBS; Christiane S. Hampe, PhD; and Anita H. Clayton, MD
Dr. Yarlagadda is with the Department of Defense and the University of Virginia in Charlottesville, Virginia. Dr. Acharya is with the Karolinska Institutet in Solna, Sweden. Dr. Kasaraneni is with Texas Tech University in Lubbock, Texas. Dr. Hampe is with the University of Washington in Seattle, Washington. Dr. Clayton is with the University of Virginia in Charlottesville, Virginia.
Funding: No funding was provided.
Disclosures: The authors have no conflicts of interest relevant to the content of this article.
Abstract: The inverse relationship between prolactin and dopamine is important in the context of treatment with antipsychotic medications in men and nonpregnant women with thought disorders. Likewise, increased levels of prolactin as confirmation of recent seizure and the reciprocal levels of prolactin and dopamine in both eclampsia (seizures) and pre-eclampsia might have significant potential effects on a growing fetus. In this article, we attempt to outline the influence of these associations on autism spectrum disorders (ASDs) in children born to mothers with established diagnoses of eclampsia and/or pre-eclampsia. Our previously published paper, “Placental Barrier and Autism Spectrum Disorders: The Role of Prolactin and Dopamine on the Developing Fetal Brain,” summarized evidence for dysregulated dopamine and prolactin levels in the etiology of ASDs and suggested a possible method for assessing whether such aberrations increase the risk of ASDs. The present paper as Part 2 expands on the published data that support this theory and proposes a study design to corroborate this hypothesis.
Keywords: autism, dopamine, placenta, prolactin
Innov Clin Neurosci. 2019;16(11–12):36–39
Autism spectrum disorder (ASD) includes autistic disorder, Asperger’s, and pervasive developmental disorder (not otherwise specified). ASD is characterized by social impairments and repetitive movements, interests, and behaviors, with symptoms developing during the first two years of life. According to the Centers for Disease Control and Prevention (CDC), ASD affects as many as 1 in 59 children, with a strong male dominance.1 While the causes of ASD remain unclear, environmental influences and genetics play a role in the development of this condition. The rapid increase in autism (from 1/150 in 2002 to 1/59 in 2010) cannot be explained by genetic changes or increased awareness and diagnosis alone.
Pathophysiology
While the pathophysiology of ASD is still unclear, neurotransmitter system dysfunction, characterized by an imbalance between excitation and inhibition, with a disproportionately high level of excitation during key stages of development, is thought to be a major contributor.2 Modulations in the neurodevelopment of the frontal/prefrontal cortex and striatum are associated with neuropsychiatric disorders, including ASD.3 Specifically, an increased connectivity between the striatum and the cortex has been observed among individuals with ASD.2,4,5 We hypothesize that elevated dopamine (DA) activity during pregnancy affects neurodevelopment in the fetus and leads to the development of ASD in the child.
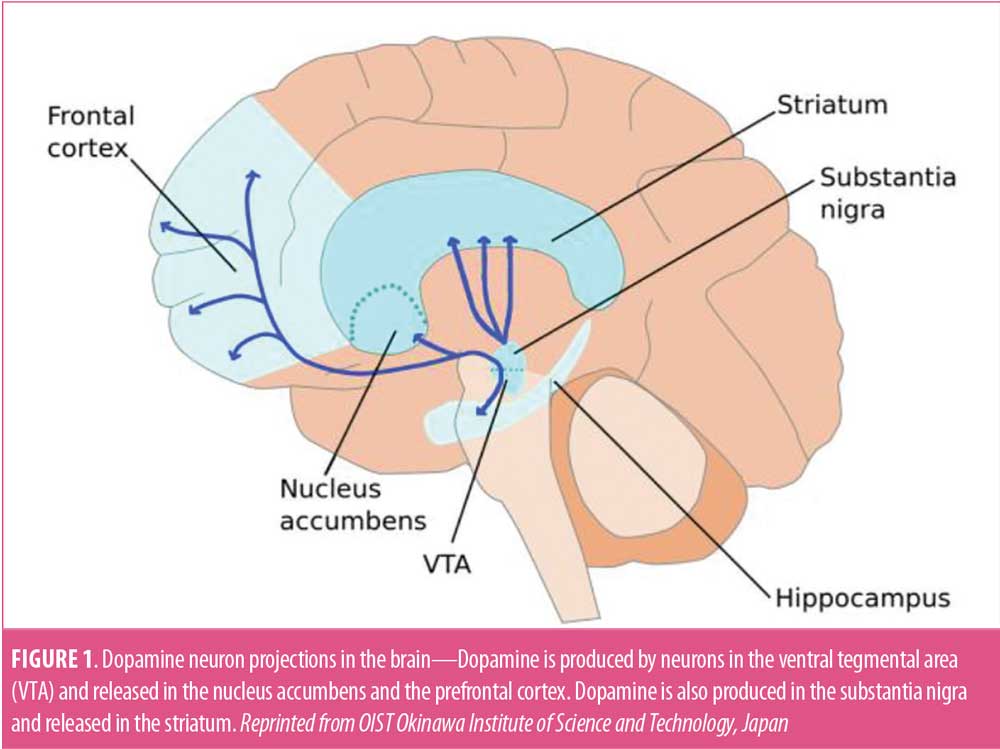
DA is an excitatory neurotransmitter with a crucial impact on early brain development.6,7 DA and its receptors appear early in neuronal development, before mature synaptic contacts form,8 and modulate developmental processes, such as neurogenesis, neuronal migration, and differentiation.9,10 Cell bodies of DA neurons are located mainly in the ventral tegmental area (VTA) and in the substantia nigra, from which they project to the striatum, the nucleus accumbens, and the prefrontal cortex, forming the mesostriatal (coordination of movement), mesolimbic (pleasure, reward), and mesocortical (motivational and emotional response) systems (Figure 1).7 Two major classes of DA receptors (D-1 like and D-2 like) are known. D1 receptor activation leads to an increase in cyclic adenosine monophosphate (cAMP), while activation of the D2 receptor leads to a decrease in cAMP levels. D1 receptors play important roles in learning and memory, locomotor activity, and reward mechanisms. While both DA receptors can be found on neurons in the cortex, striatum, and nucleus accumbens, DA neurons themselves only express D2 receptors (autoreceptors), so that the release of DA provides negative feedback regulation. Dysbalance in the DA system has been associated with neurological disorders of developmental origin including ASD.6
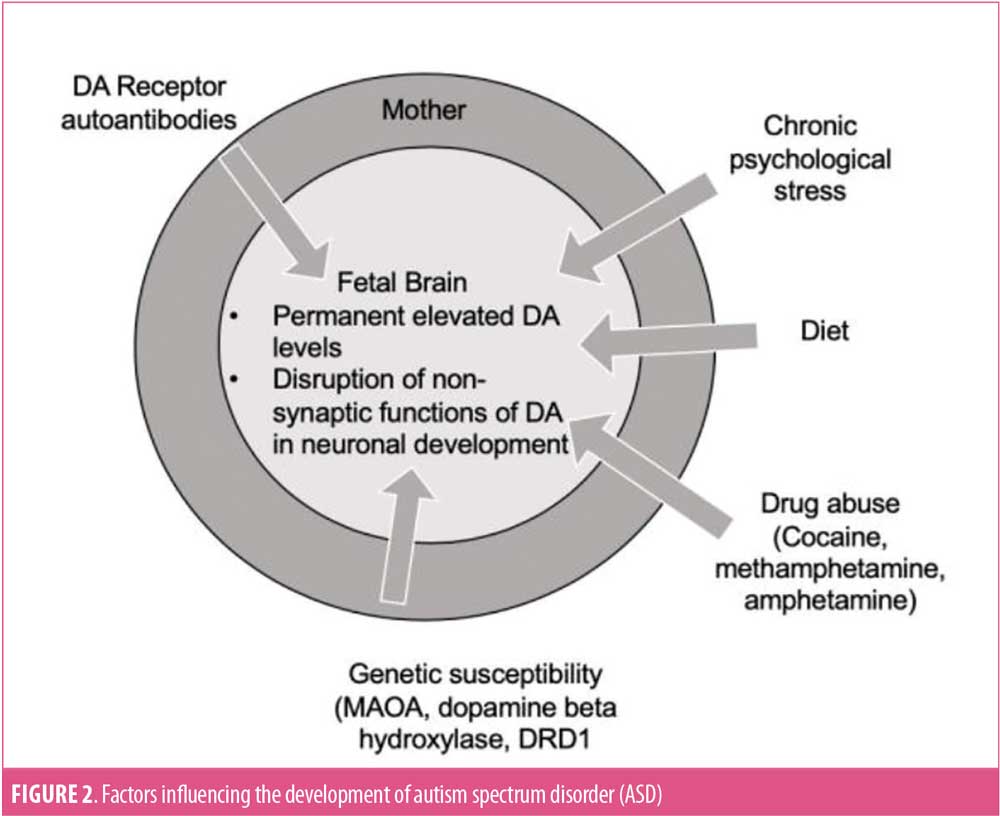
Fetal innervation and receptor expression can be affected through direct and indirect manipulations of maternal DA levels during pregnancy. Manipulating factors, including drug abuse,11 diet,12 maternal hyperthermia, maternal psychosocial stress,13 hypoxia,14 environmental neurotoxins,15 and immune challenge,16 are associated with altered DA activity in developing offspring and might influence learning behaviors and motoric activity. The two perinatal factors most consistently linked to autism—cesarean birth and low APGAR scores at birth—are both associated with elevated DA transmission in the newborn (Figure 2).
The hypothesis that an overactive DA system is involved in brain dysfunction in autism is supported by observations in humans, including:
- Beneficial effect of D2 receptor antagonists on some aspects of autism.17
- Increased striatal D2 receptor-binding in children with autism.18
- Hyperfunctional DA systems in the medial region of the orbitofrontral cortex of children with autism.19
- Elevated DA synthesis and storage in the striatum and frontal cortex of adults with Asperger’s syndrome.20
- Increased levels of the principal DA metabolite, homovanillic acid (HVA) in the cerebrospinal fluid (CSF),21,22 and urine23 of individuals with autism.
More evidence supporting our hypothesis that high prenatal DA levels produce lasting neurochemical and behavioral changes comes from animal studies:
- DA knockout mice are severely hypoactive,24 while DA transporter (DAT) knockdown and knockout mice are hyper-DArgic and display novelty-induced hyperactivity.25,26
- The administration of DA in DA-deficient mouse models induces hyperactivity.27
- The administration of L-DOPA to pregnant mice leads to elevated DA concentrations in the embryonic brain. The adult offspring of L-DOPA-treated mothers show persisting elevations in DA levels of the striatum and midbrain, with significantly increased basal DA release in the nucleus accumbens. Moreover, the animals show significantly lower cocaine-conditioned place preference, a measure of DA-mediated behavior.28
Anti-DA Receptor Autoantibodies
DA activity might also be impacted by the presence of activating anti-DA receptor antibodies. The notion of pathogenic DA receptor autoantibodies in neurological disorders dates back to findings in the late 1970s and early 1980s29,30 and is currently being investigated in relation to pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections.31 Autoantibodies against the D2 receptors were reported in Sydenham chorea32,33 and a small study revealed the presence of D2R autoantibodies in 30 percent of patients with Sydenham chorea and 10 percent of patients with Tourette’s syndrome, while none of the control subjects tested positive. In Tourette’s syndrome, the presence of D2R autoantibodies was significantly higher among female patients (40%) than male patients (5%).34
Individuals with ASD have increased rates of family members with a history of autoimmune disease.35,36 Recent studies have shown that 10 percent of mothers of a child with ASD test positive for autoantibodies reactive to the fetal brain.37,38 These autoantibodies react to antigens in the frontal cortex, hippocampus, and cerebellum.38 So far, seven candidate autoantigens, including lactate dehydrogenase A and B, stress-induced phosphoprotein 1, collapsing response mediator proteins 1 and 2, cypin, and Y-box binding protein have been identified.39
It is well-known that maternal antibodies cross the placenta and might affect fetal brain development since the fetal blood–brain barrier is not fully formed.40 To test the potential pathogenicity of these autoantibodies, pregnant mice or monkeys received autoantibodies from mothers of an ASD child. Behavioral changes in the offspring suggest a pathogenic role.41,42
Plan for Prospective Study
Description of specific aims and analytic approach. We hypothesize that high DA activity during pregnancy can be associated with the development of ASD in the offspring. Elevated DA activity can be caused by a number of mechanisms, including increased production, decreased breakdown,43 decreased re-uptake,44 and/or activation of DA receptor autoantibodies.32,33
To test our hypothesis, we will correlate DA activity (directly or through surrogate markers, see below) in mothers with a child with ASD during a subsequent pregnancy with that in cord blood and in the child (at 1 year and at diagnosis of ASD).
Determine central nervous system (CNS) DA levels during pregnancy (indirect), cord blood (direct), and in samples obtained from offspring (indirect).
Determine titers of autoantibodies against D1 and D2 DA receptors in the mothers during early, mid-, and late gestation, cord blood, and in samples obtained from offspring.
To test our hypothesis that elevated DA activity during pregnancy is a risk factor for ASD in the child, we will compare maternal and cord blood samples between children who developed ASD and those who remained healthy. If these results are positive, DA activity could be used as a biomarker for ASD risk.
Finally, to test our hypothesis that maternal DA activity is transferred to the child and processes throughout early childhood, we will compare DA activity during pregnancy with DA activity in the newborn and in the child at diagnosis of ASD. Such a correlation between maternal and neonatal DA activity is expected based on previous studies.45
Description of data/samples is needed (including discussion of timing of data/sample transfer and a discussion of specific points described above regarding biosamples).
For a complete analysis of HVA and prolactin (indirect measure of central DA level) and the determination of autoantibodies directed against D1 and/or D2 receptors, a minimum of 750µL of plasma or serum is required, including the following:
- 500µL of plasma/serum for HVA liquid chromatography with tandem mass spectrometry (mothers and offspring)
- 200µL of plasma/serum for chemiluminescence prolactin method (mothers and children)
- 50µL of plasma/serum for antibody testing (mothers and children)
Measurement of central DA levels. DA does not cross the blood–brain barrier, and central DA levels can therefore not be directly determined without an invasive spinal tap. However, several peripheral markers have been used to indirectly determine central DA levels. One of these is prolactin, a hormone that is predominately produced by the anterior pituitary gland.46–48 Prolactin secretion from the pituitary gland is mainly regulated by DA, which inhibits prolactin secretion. Prolactin levels increase during pregnancy 10- to 20-fold.
Decreased baseline levels of prolactin were found in patients with autism,49 suggesting increased central DA levels. However, prolactin is also produced in the hypothalamus, where its secretion is stimulated by serotonin.50 Therefore, low prolactin levels might be due to diminished serotonin function, increased activity of DA, or both.
Another marker is the presence of HVA in urine and serum.51,52 HVA is a metabolite of DA; however, only 25 percent of urine and plasma HVA is the result of central DA turnover, so that plasma HVA might only be useful in determining substantial changes in central DA activity.53
A study of 156 children with autism measured urine levels of DA and HVA and showed significantly lower levels of DA and HVA in medicated children with autism.23
Peripheral prolactin and HVA levels are currently the preferred tests to determine central DA concentrations. HVA levels in urine or serum are quantitatively determined by tandem mass spectrometry while prolactin levels in serum or plasma are quantitatively determined by chemiluminescence.
A larger analysis intended to generate preliminary data will include a description of how the preliminary data generated will be used for subsequent grant applications.
Preliminary data will be prepared for publication in manuscript form and presentations at scientific meetings. Preliminary data will be used for subsequent funding request from the National Institutes of Health.
References
- Baio J, Wiggins L, Christensen DL, et al. Prevalence of autism spectrum disorder among children aged 8 years—autism and developmental disabilities monitoring network, 11 sites, United States, 2014. MMWR Surveill Summ. 2018;67(6):1–23.
- Rubenstein J, Merzenich M. Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2003;2(5):255–267.
- Langen M, Leemans A, Johnston P, et al. Fronto-striatal circuitry and inhibitory control in autism: findings from diffusion tensor imaging tractography. Cortex. 2012;48(2):183–193.
- Delmonte S, Gallagher L, O’Hanlon E, et al. Functional and structural connectivity of frontostriatal circuitry in autism spectrum disorder. Front Hum Neurosci. 2013;7:430.
- Di Martino A, Kelly C, Grzadzinski R, et al. Aberrant striatal functional connectivity in children with autism. Biol Psychiatry. 2011;69(9):847–856.
- Turner K, Frost L, Linsenbardt D, et al. Atypically diffuse functional connectivity between caudate nuclei and cerebral cortex in autism. Behav Brain Funct. 2006;2:34.
- Money K, Stanwood G. Developmental origins of brain disorders: roles for dopamine. Front Cell Neurosci. 2013;7:260.
- Bjorklund A, Dunnett S. Dopamine neuron systems in the brain: an update. Trends Neurosci. 2007;30(5):194–202.
- Araki K, Sims J, Bhide P. Dopamine receptor mRNA and protein expression in the mouse corpus striatum and cerebral cortex during pre- and postnatal development. Brain Res. 2007;1156:31–45.
- Bhide P. Dopamine, cocaine and the development of cerebral cortical cytoarchitecture: a review of current concepts. Semin Cell Dev Biol. 2009;20(4):395–402.
- Frederick A, Stanwood G. Drugs, biogenic amine targets and the developing brain. Dev Neurosci. 2009;31(1-2):7–22.
- Kubrusly R, Bhide P. Cocaine exposure modulates dopamine and adenosine signaling in the fetal brain. Neuropharmacology. 2010;58(2):436–443.
- Kuperstein F, Eilam R, Yavin E. Altered expression of key dopaminergic regulatory proteins in the postnatal brain following perinatal n-3 fatty acid dietary deficiency. J Neurochem, 2008;106(2):662–671.
- Son G, Chung S, Geum D, et al. Hyperactivity and alteration of the midbrain dopaminergic system in maternally stressed male mice offspring. Biochem Biophys Res Commun. 2007;353(3):823–829.
- Graf A, Maslova M, Trofimova L, et al. Effect of antenatal hypoxia on age-specific dynamics of ECG parameters and content of biogenic amines in the central nervous system. Bull Exp Biol Med. 2007;144(2):188–191.
- Fonnum F, Mariussen E. Mechanisms involved in the neurotoxic effects of environmental toxicants such as polychlorinated biphenyls and brominated flame retardants. J Neurochem. 2009;111(6):1327–1347.
- Meyer U, Engler A, Weber L, et al. Preliminary evidence for a modulation of fetal dopaminergic development by maternal immune activation during pregnancy. Neuroscience. 2008;154(2):701–709.
- McDougle C, Erickson C, Stigler K, Posey D. Neurochemistry in the pathophysiology of autism. J Clin Psychiatry. 2005;66 Suppl 10:9–18.
- Fernell E, Watanabe Y, Adolfsson I, et al. Possible effects of tetrahydrobiopterin treatment in six children with autism—clinical and positron emission tomography data: a pilot study. Dev Med Child Neurol. 1997;39(5):313–318.
- Nakamura K, Sekine Y, Ouchi Y, et al. Brain serotonin and dopamine transporter bindings in adults with high-functioning autism. Arch Gen Psychiatry. 2010;67(1):59–68.
- Nieminen-von Wendt T, Metsähonkala L, Kulomäki T, et al. Changes in cerebral blood flow in Asberger syndrome during theory of mind tasks presented by the auditory route. Eur Child Adolesc Psychiatry. 2003;12(4):178–189.
- Gillberg C, Svennerholm L, Hamilton-Hellberg C. Childhood psychosis and monoamine metabolites in spinal fluid. J Autism Dev Diord. 1983;13(4):383–396.
- Gillberg C, Svennerholm L. CSF monoamines in autistic syndromes and other pervasive developmental disorders of early childhood. Br J Psychiatry. 1987;252:89–94.
- Martineau J, Herault J, Petit E, et al. Catecholaminergic metabolism and autism. Dev Med Child Neurol. 1994;36(8):688–697.
- Zhou Q, Palmiter R. Dopamine-deficient mice are severely hypoactive, adipsic, and aphagic. Cell. 1995;86(7):1197–1209.
- Zhuang X, Oosting R, Jones S, et al. Hyperactivity and impaired response habituation in hyperdopaminergic mice. Proc Natl Acad Sci USA. 2001;98(4):1982–1987.
- Gainetdinov R, Wetsel W, Jones S, et al. Role of serotonin in the paradoxical calming effect of psychostimulants on hyperactivity. Science. 1999;283(5400):397–401.
- Chartoff E, Marck B, Matsumoto A, et al. Induction of stereotypy in dopamine-deficient mice requires striatal D1 receptor activiation. Proc Natl Acad Sci USA. 2001;98(18):10451–10456.
- Ren J, Jiang Y, Wang Z, et al. Prenatal L-DOPA exposure produces lasting changes in brain dopamine content, cocaine-induced dopamine release and cocaine conditioned place preference. Neuropharmacology. 2011;60(2–3):295–302.
- Knight J. Dopamine-receptor-stimulating autoantibodies: a possible cause of schizophrenia. Lancet. 1982;2(8307):1073–1076.
- Abramsky O, Litvin Y. Autoimmune response to dopamine-receptor as a possible mechanism in the pathogenesis of Parkinson’s disease and schizophrenia. Perspect Biol Med. 1978;22(1):104–114.
- Cunningham M, Cox C. Autoimmunity against dopamine receptors in neuropsychiatric and movement disorders: a review of Sydenham chorea and beyond. Acta Physiol (Oxf). 2016;216(1):90–100.
- Cox C, Sharma M, Leckman J, et al. Brain human monoclonal autoantibody from sydenham chorea targets dopaminergic neurons in transgenic mice and signals dopamine D2 receptor: implications in human disease. J Immunol. 2013;191(11):5524–5541.
- Ben-Pazi H, Stoner J, Cunningham M. Dopamine receptor autoantibodies correlate with symptoms in Sydenham’s chorea. PloS One. 2013;8(9):e73516.
- Dale R, Merheb V, Pillai S, et al. Antibodies to surface dopamine-2 receptor in autoimmune movement and psychiatric disorders. Brain. 2012;135(pt11):3453–3468.
- Altadóttir H, Pedersen M, Thorsen P, et al. Association of family history of autoimmune diseases and autism spectrum disorders. Pediatrics. 2009;124(2):687–694.
- Keil A, Daniels J, Forssen U, et al. Parental autoimmune diseases associated with autism spectrum disorders in offspring. Epidemiology. 2010;21(6):805–808.
- Croen L, Braunschweig D, Haapanen L, et al. Maternal mid-pregnancy autoantibodies to fetal brain protein: the early markers for autism study. Biol Psychiatry. 2008;64(7):583–588.
- Brimberg L, Sadiq A, Gregersen P, Diamond B. Brain-reactive IgC correlates with autoimmunity in mothers of a child with an autism spectrum disorder. Mol Psychiatry. 2013;18(11):1171–1177.
- Braunschweig D, Krakowiak P, Duncanson P, et al. Autism-specific maternal autoantibodies recognize critical proteins in developing brain. Transl Psychiatry. 2013;3:e277.
- Saunders N, Liddelow S, Dziegielewska K. Barrier mechanisms in the developing brain. Front Pharmacol. 2012;3:46.
- Singer H, Morris C, Gause C, et al. Prenatal exposure to antibodies from mothers of children with autism produces neurobehavioral alterations: a pregnant dam mouse model. J Neuroimmunol. 2009;211(1–2):39–48.
- Martin L, Ashwood P, Braunschweig D, et al. Stereotypies and hyperactivity in rhesus monkeys exposed to IgG from mothers of children with autism. Brain Behav Immun. 2008;22(6):806–816.
- Robinson P, Schutz C, Macciardi F, et al. Genetically determined low maternal serum dopamine beta-hydroxylase levels and the etiology of autism spectrum disorders. Am J Med Genet. 2001;100(1):30–36.
- Hamilton P, Campbell N, Sharma S, et al. De novo mutation in the dopamine transporter gene associates dopamine dysfunction with autism spectrum disorder. Mol Psychiatry. 2013;18(12):1315–1323.
- Tuomisto J, Mannisto P. Neurotransmitter regulation of anterior pituitary hormones. Pharmacol Rev. 1985;37(3):249–332.
- Bridge M, Weller A, Rayson M, Jones D. Ambient temperature and the pituitary hormone responses to exercise in humans. Exp Physiol. 2003;88(5):627–635.
- Freeman M, Kanyicska B, Lerant A, Nagy G. Prolactin: structure, function, and regulation of secretion. Physiol Rev. 2000;80(4):1523–1631.
- Hoshino Y, Yamamoto T, Kaneko M, et al. Blood serotonin and free tryptophan concentration in autistic children. Neuropsychobiology. 1984;11(1):22–27.
- Maas J, Hattox S, Greene N, Landis D. Estimates of dopamine and serotonin synthesis by the awake human brain. J Neurochem. 1980;34(6):1547–1549.
- Field T, Diego M, Hernandez-Reif M, et al. Prenatal dopamine and neonatal behavior and biochemistry. Infant Behav Dev. 2008;31(4):590–593.
- Lambert G, Eisenhofer G, Cox H, et al. Direct determination of homovanillic acid release from the human brain, an indicator of central dopaminergic activity. Life Sci. 1991;49(15):1061–1072.
- Kopin I, White J, Bankiewicz K. A new approach to biochemical evaluation of brain dopamine metabolism. Cell Mol Neurobiol. 1988;8(20):171–179.