

by Gahan J. Pandina, PhD; Joan Busner, PhD; Lucas Kempf, MD; Joan Fallon, BS, DC; Larry D. Alphs, MD, PhD; Maria T. Acosta, MD; Anna-Karin Berger, PhD; Simon Day, PhD; Judith Dunn, PhD; Victoria Villalta-Gil, PhD; Margaret C. Grabb, PhD; Joseph P. Horrigan, MD; William Jacobson, PhD; Judith C. Kando, PharmD, BCPP; Thomas A. Macek, PharmD, PhD; Manpreet K. Singh, MS, MD; Arielle D. Stanford, MD; and Silvia Zaragoza Domingo, PhD
All authors are members of the International Society for Central Nervous System Clinical Trials and Methodology (ISCTM) Working Group for Rare Disease/Orphan Drug Development; Drs. Pandina and Busner are Co-Chairs. Dr. Pandina is with Janssen Research & Development in Titusville, New Jersey. Dr. Busner is with Signant Health in Blue Bell, Pennsylvania and Department of Psychiatry, Virginia Commonwealth University School of Medicine in Richmond, Virginia. Dr. Kempf is with Parexel in Washington, DC. Dr. Fallon is with Curemark in Rye Brook, New York. Dr. Alphs is with Denovo Pharmaceuticals in Princeton, New Jersey. Dr. Acosta is with the National Institutes of Health in Bethesda, Maryland. Dr. Berger is with H. Lundbeck A/S in Valby, Denmark. Dr. Day is with Clinical Trials Consulting & Training in Buckingham, United Kingdom. Dr. Dunn is with Evolution Research Group in Boston, Massachusetts. Dr. Villalta-Gil is with WCG Clinical in Durham, North Carolina. Dr. Grabb is with the National Institute of Mental Health in Rockville, Maryland. Dr. Horrigan is with AMO Pharma in Wonersh, United Kingdom and Duke University in Durham, North Carolina. Dr. Jacobson is with Harmony Biosciences in Mundelein, Illinois. Dr. Kando is with Karuna Therapeutics in Boston, Massachusetts. Dr. Macek is with Novartis Pharmaceuticals in Bannockburn, Illinois. Dr. Singh is with Stanford University School of Medicine in Stanford, California. Dr. Stanford is with Bristol Myers Squibb in Cambridge, Massachusetts. Dr. Domingo is with Neuropsynchro in Barcelona, Spain.
Funding: No funding was provided for this article.
Disclosures: GJP is a full-time employee of Janssen Research & Development, LLC, and a Johnson & Johnson stockholder. JB is a full time employee of Signant Health, and may own stock or equity in the company. LDH holds stock in Johnson & Johnson. JPH is a shareholder of AMO Pharma, Ltd. JCK holds stock in McKesson, Johnson & Johnson, and Takeda. TAM is an employee and shareholder of Novartis Pharmaceuticals. MKS has received research support from Stanford’s Maternal Child Health Research Institute and Stanford’s Department of Psychiatry and Behavioral Sciences, National Institute of Mental Health, National Institute of Aging, Patient Centered Outcomes Research Institute, Johnson & Johnson, and the Brain and Behavior Research Foundation; is on a data safety monitoring board for a study funded by the National Institute of Mental Health; is on the advisory board for Sunovion and Skyland Trail; is a consultant for Johnson & Johnson, Alkermes, Neuroma, AbbVie, Karuna Therapeutics, Inc., Boehringer-Ingelheim, Intra-Cellular Therapeutics, Inc, and Alto Neuroscience; and receives honoraria from the American Academy of Child and Adolescent Psychiatry, royalties from American Psychiatric Association Publishing and Thrive Global, and travel support for continuing medical education from Neuroscience Education Institute and Psych Congress. SZD has provided services as consultant during the last two years to Medavante-Prophase and Sanofi. All other authors conflicts of interest relevant to the content of this article.
Innov Clin Neurosci. 2024;21(1–3):52–60.
Abstract
The 1983 Orphan Drug Act in the United States (US) changed the landscape for development of therapeutics for rare or orphan diseases, which collectively affect approximately 300 million people worldwide, half of whom are children. The act has undoubtedly accelerated drug development for orphan diseases, with over 6,400 orphan drug applications submitted to the US Food and Drug Administration (FDA) from 1983 to 2023, including 350 drugs approved for over 420 indications. Drug development in this population is a global and collaborative endeavor. This position paper of the International Society for Central Nervous System Clinical Trials and Methodology (ISCTM) describes some potential best practices for the involvement of key stakeholder feedback in the drug development process. Stakeholders include advocacy groups, patients and caregivers with lived experience, public and private research institutions (including academia and pharmaceutical companies), treating clinicians, and funders (including the government and independent foundations). The authors articulate the challenges of drug development in orphan diseases and propose methods to address them. Challenges range from the poor understanding of disease history to development of endpoints, targets, and clinical trials designs, to finding solutions to competing research priorities by involved parties.
Keywords: Orphan disease, key stakeholders, patient experience, clinical trials, drug development research
The development of the 1983 Orphan Drug Act in the United States (US) changed the landscape of the development of therapeutics for rare disorders. Authored by Representative Harry Waxman, passed by Congress, and signed into law by President Ronald Reagan, the Orphan Drug Act attempted to bring attention to and help resolve difficulties with drug development for patients with rare diseases. For most rare diseases, clinical trials are highly complex and costly, with considerable regulatory challenges. Although addressing unmet medical need is of primary interest, a complication is that this complexity offers little hope of fair and reasonable financial returns for sponsors and investors. Lacking an operational definition, the Orphan Drug Act was amended in 1984 to provide a definition of an orphan drug as a drug or treatment for a rare disease that affects fewer than 200,000 people in the US and for those diseases with more than 200,000 affected but where there is little hope of recouping costs for getting the drug to market.
Although individually rare, orphan diseases collectively affect approximately 300 million people worldwide, half of whom are children.1 One-third of individuals with orphan diseases are likely to die by the age of five years.2 With recent research advances, it is now known that 80 percent of orphan diseases are genetic disorders, and 95 percent have no current treatments.3 The gap between increasing scientific knowledge and poor outcomes is influenced by the lack of precedence for developing treatments, along with incomplete knowledge of the natural history of many of these disorders.
A common measure of the success of the Orphan Drug Act since its enactment is the increasing number of new treatments approved for rare diseases. From 1983 to 2023, over 6,400 orphan drug applications were submitted to the US Food and Drug Administration (FDA), with the approval of approximately 350 drugs for over 420 indications; comparatively, only 34 drugs were eligible for approval in the period between 1967 and 1983.4,5 The Orphan Drug Act has been widely credited for accelerating the growth of novel therapeutics in this area, along with the molecular biology revolution and the Human Genome Project. Many important treatment advances have occurred in 2022 and 2023 alone.6
With over 7,000 rare disease types identified worldwide, rare diseases are referred to in some regions as significant minorities. The research on orphan drugs and the policies adopted by the US Congress have spurred similar acts in multiple countries and territories, such as Japan, Europe, Singapore, Australia, Taiwan, and South Korea. In total, 92 countries/regions have legislation, regulations, or policies that in some way facilitate patient access to orphan drugs.5
However, the rules and incentives around orphan drug development vary widely from one jurisdiction to another. This is in no small part due to the differing levels of grassroots efforts to encourage drug development by advocacy groups and other concerned parties. Drug development in rare diseases, however, is seen as a global and collaborative endeavor due to the scarcity of patients, knowledge, resources, and expert clinical investigators at research centers.
Advocacy
The Huntington’s Disease Society of America, (founded in 1967), was one of the first advocacy groups to interact with the US Congress prior to the passage of the Orphan Drug Act. In the years that followed, multiple advocacy groups have encouraged or urged academics, sponsors, and the FDA to approve new treatments for unmet medical needs in the rare disease area. Dunkle et al7 described the growth of patient advocacy groups and their relationship to government research, funding, and regulatory entities as a critical component to derisking orphan product development. Many of the initiatives started by advocacy groups before and shortly after the Orphan Drug Act continue today through strategic research initiatives, such as patient registries, obtaining natural histories of diseases, and vocal advocacy for the development of effective and safe interventions. Today, many of these initiatives have become global.
To promote changes in rare disease regulatory policies, aggregate organizations began to form, such as the National Organization for Rare Diseases (NORD), EURODIS, Global Genes, or Faster Cures, which provide varying levels of local and national advocacy. NORD was formed in 19838 as an ad hoc coalition of leaders in the rare disease space to bring attention to rare diseases, which helped with the creation of the Orphan Drug Act. NORD currently serves as the US hub of information for patients, families, researchers, sponsors, and other organizations serving those with rare diseases. Other national and international organizations have followed this example, building on this effort globally or with different educational, policy, research, or diagnostic emphases, resulting in multiple partnerships.9 The International Rare Diseases Research Consortium (IRDRC),10 for example, not only facilitates the development of new therapies, but also focuses on early diagnosis and intervention, with the following vision: to “[e]nable all people living with a rare disease to receive an accurate diagnosis, care, and available therapy within one year of coming to medical attention.”
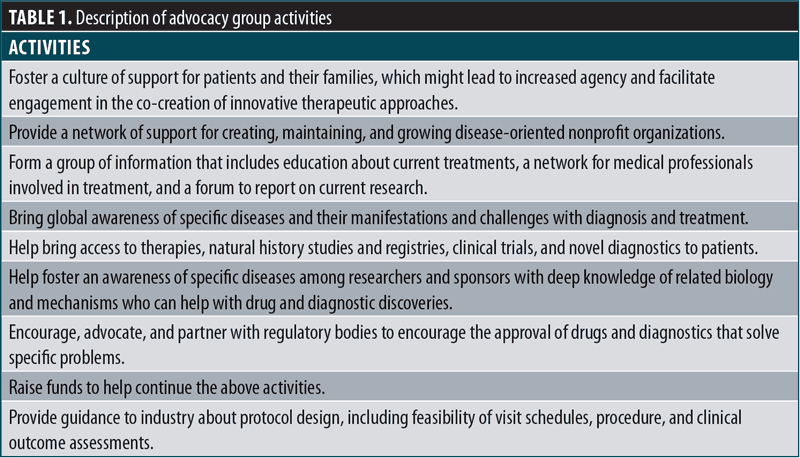
Despite varying weights of emphasis, the overarching, multifold focus of these advocacy groups is strong and continues to deliver impact for relevant stakeholders. Table 1 provides an overview of many of the ways in which advocacy groups advance novel therapeutic development of orphan drugs.
Articulating Challenges in Rare Disease Drug Development in Order to Find Ways to Address Them
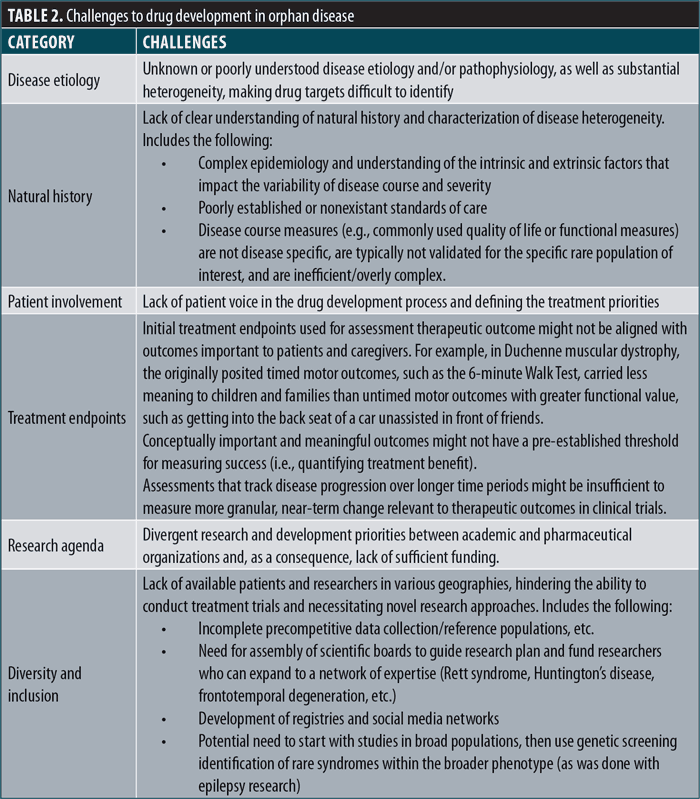
Certain decision makers, including scientists and patient advocacy groups in the rare disease space, have recently begun to identify the challenges associated with orphan drug development. As a result, some increase in potential financial incentives for orphan drug development globally has been noted, although a multiplicity of issues remains. Table 2 provides broad categories and specific challenges facing drug development in orphan disease.
As we approach the 40th anniversary of the 1983 Orphan Drug Act, we can use the accumulated data, research methods, and experience to inform best practices moving into the future.
In the following sections, we examine the critical role of advocacy group partnership in addressing the potential challenges for drug discovery for central nervous system (CNS) orphan diseases, as well as what has and will inform current and future best practices.
Disease etiology: unknown or poorly characterized disease etiology and/or pathophysiology. Some rare diseases are so rare that there has been little to no research into their etiology or pathophysiology. Genetic research provides interesting hypotheses about neurobiological pathways that might lead to potential drug targets.11,12 However, animal models that extensively study the underlying mechanisms of rare diseases and therapeutic strategies have inherent limitations due to genetic heterogeneity with human models. Recent innovations in artificial intelligence (AI) and in vivo models using brain organoids and brain-on-a-chip technologies might augment efforts in what remains a highly challenging area for drug discovery.13,14 Nevertheless, early in the identification phase of rare diseases, stakeholders can more effectively connect with the limited number of dispersed academic labs in early research and disease identification. This could be done via support groups, rare/orphan disease societies, governmental research groups, or other mechanisms.
Natural history: lack of clear understanding of natural history and characterization of disease heterogeneity. Prior to the Orphan Drug Act, very little focus was placed on gathering information about diagnosis, natural history, disease burden, standards of care, or other important disease information necessary for drug development. Advocacy groups mostly create registries limited to sociodemographic data and lists of physicians with some experience in treating the disease. Early registries, social network groups, and groups such as PatientsLikeMe evolved to aggregate clinical data on the natural history and course of disease, early diagnosis, and development of clinical endpoints and biomarkers. These and related data help inform physicians, as well as researchers, who can then use aggregated data for trial planning, study design, and endpoint validation.
Data collection is also limited, and might never be available, due to the lack of validated diagnostic and outcome assessments that could help track the course and progression of illness and treatment response.15 Furthermore, the cataloging of the natural course of the diseases is beyond the scope of the clinical trial setting, so this important work must be done in other settings.
The lack of true incidence and prevalence data on orphan diseases, particularly those with heterogenous presentations, early in the course of identifying the disease are key negative factors in facilitating therapeutics for rare diseases. The true epidemiology of rare diseases is often poorly understood for years; without proper, early diagnosis, information might be lacking in healthcare databases or via government agency sources. The global nature of disease presentation, the course of a specific illness, and regional standards of care are typically unknown and often remain poorly characterized until advocacy groups help to aggregate information. Regional differences are important factors not only in finding and treating patients, but in identifying the intrinsic and extrinsic factors that play a role in the heterogeneity of the disease presentations and clinical course. Potential confounds need to be controlled for in global clinical trials. In addition, eliciting patient and caregiver input on the regional variations in diagnosis, standards of care, and even clinical significance and interpretation of endpoints are key to developing global clinical trial designs. For example, while it is common knowledge that access to antiepileptic drugs varies widely across the globe, regional variations in cultural understanding and interpretation of rare forms of epilepsy are not well understood, and commonly used endpoints might not be culturally adapted. The general lack of early disease knowledge also leads to a lack of early validated, predictive screening procedures, further adding to the challenges of early diagnosis and proper epidemiologic classification. For example, physicians do not typically screen for Rett syndrome in newborn babies,16 leading to delayed identification, diagnosis, and treatment; in contrast, spinal muscular atrophy (SMA) has a common, inheritable mutation, which has led to more prevalent screening.17
Patient involvement: lack of patient/caregiver input in drug development process and priorities. The goal of drug development is to bring to approval safe and effective therapies that address important unmet medical needs. To accomplish this goal, it is critical to gain insights on the ultimate end user: the patient. In some rare diseases and due to cognitive or other impairments (e.g., speech/communication), it might not be possible to obtain direct input from the patient, so input on unmet medical needs is solicited from caregivers and family members. Many patients are consistently able to articulate what they find most meaningful in terms of a positive therapeutic response, the most troubling or disabling symptoms they experience, and what they are willing to tolerate in terms of side effect burden. Gaining this perspective early on can help guide a clinical development program that ultimately is able to identify a therapeutic intervention that provides demonstrable real-world benefit with a tolerable side effect profile. Where patients are unable to voice their own concerns, such as with nonverbal or very young pediatric populations, every effort should be made to include primary caregivers of these patients as a required group so that these perspectives can be included. Caregivers can also provide longitudinal perspective and information from other treatment providers that might not be as easily gleaned from patients.
There are many ways to obtain patient input. It has been suggested that structured quantitative patient research, built on robust and systematically gathered qualitative insights, should be utilized early on and throughout the development process.18 This can be done thorough surveys, patient interviews and focus groups, for example. Another way to gather insights is through social media, as patients and caregivers often post information on Facebook, X (formerly Twitter), YouTube, online bulletin boards, and blogs. In addition, surveys, semistructured one-on-one interviews, and patient advisory boards might also prove helpful. Information gleaned from these less formal sources will inform decision making to assure that research is conducted in the best interest of the patient and generate hypotheses to be confirmed by other, more reliable and comprehensive methods. This information can be used to guide product design, delivery systems, target symptoms, and tolerable side effect profiles, as well as many other aspects of outcome. This knowledge can also inform regulatory and payer strategies by keeping the treatment outcome focus patient-centric.
Treatment endpoints: not patient-centered or aligned to outcomes important to patients and caregivers. Lack of inclusion of patients and caregivers in early research, lack of disease understanding/heterogeneity, and limited funding early in disease identification leads to poor consensus on important treatment targets and clinical outcomes of interest. These factors also confound the development of reliable and valid outcome measures, resulting in measures of convenience being utilized for early clinical trials.
Industry sponsors often need to work with key stakeholders to target therapeutic outcomes that are acceptable to regulatory agencies in the absence of full knowledge of disease causation or known neurobiological etiology. When etiology is known (i.e., specific, known genetic abnormality with associated neurobiology), real-world data can help inform researchers and regulatory authorities of clinical outcomes that matter most to patients and families. The struggle to identify clinically meaningful outcome measures that mirror or reflect the nature of a rare disease proves difficult even today. Differences in priorities and outcome needs remain among patients/caregivers, regulators, and trial sponsors.
Patients and caregivers are looking for solutions to solve problems affecting many or all aspects of daily life. The need to treat life-threatening symptoms or ease the burden of care are common foci of interest. Regulators might look for outcome measures that improve a disease based on mechanism of action (i.e., objective markers of biology or causation), but in rare diseases, these mechanisms might not yet be identified or might be difficult to target therapeutically.
Sponsors are often caught between these two extremes, as is the case with the recently approved drug for Duchenne muscular dystrophy (DMD). DMD is a genetic disorder that causes alterations in the dystrophin protein, resulting in muscle cell degeneration and fatty replacement. This disease primarily affects male patients and can present as early as two years of age. It progressively worsens, such that patients are often confined to a wheelchair in their late teens and twenties, followed by premature death. Delandistrogene moxeparvovec-rokl, the first approved therapeutic for the treatment of DMD, was approved against the advice of the FDA expert panel. Objections to the approval were based on concerns regarding the primary outcome measure used in the clinical trials conducted for drug approval (the 6-minute Walk Test), a lack of placebo arm, and the use of a small sample size of six subjects in the evaluation of the drug. Furthermore, the FDA was looking for an outcome measure that related to elevating levels of dystrophin.19 In this landmark case in which FDA drug approval occurred against the advice of the advisors, the sponsor and patients were extremely happy, and the regulators faced split opinions from many public sources.
Benchmarking between more fit-for-purpose endpoints and patient-centric outcomes is needed through strategic research years in advance of the development of novel drugs.
Research agenda in rare diseases: divergent research and development priorities between academic and pharmaceutical organizations and lack of funding. Academic research, wherein potential disease targets are identified, relevant biomarkers are discovered, and disease characteristics and pathologies are uncovered, is important in the early discovery phase of biomedical research and treatment development. These discoveries help to inform and build a foundation for pharmaceutical drug development programs, which are then typically carried out by private industry or in public-private partnerships. In the case of rare diseases, however, pharmaceutical companies are beginning to become more involved in drug development; their involvement was limited until recently due to both the lack of a clear path to indication and low expectation of a reasonable financial return. Academic investigators, through investigator-sponsored studies, might serve to establish a pathway for a translational research drug development program, including the testing of novel, disorder-specific outcome measures that fill a research need. While industry sponsors might have the expertise to develop these measures within a company, they intentionally rely on expert consensus opinion and guidelines to establish what are valid and reliable outcomes related to diseases of interest.
There are early scientific considerations and go/no-go decision points in drug development that might not be clear to academic investigators as products move from target identification, compound screening, optimization, and Investigational New Drug (IND)-enabling studies. These commonly include complex and difficult-to-address questions raised by institutional review boards (IRBs) and regulatory authorities with regard to the disease state. Examples of considerations commonly faced within a company during the drug development process include protecting intellectual property; expertise in medicinal chemistry, pharmacology, toxicology, and targeted organ issues; experience in go/no-go decision making for development; and project management and operational processes. The disconnect between nonclinical, basic biological research and applied clinical research has often stymied the drug development process in rare diseases.
Foundations for rare diseases have recognized these hurdles and provided a variety of approaches to overcome them. For example, the Cystic Fibrosis Foundation has established a subsidiary nonprofit organization, Cystic Fibrosis Foundation Therapeutics, Inc. (CFFT), which enables collaborations with biopharmaceutical companies to support a broad drug development pipeline from discovery through clinical testing. This includes high-throughput assay and biomarker development, compound screening, and drug optimization. The CFFT further establishes research networks, a biospecimen bank, standardized outcome measures for use in trials, a patient registry that tracks health outcomes, and Good Clinical Practice (GCP) training for investigators and staff.20 The activities and approach taken by the CFFT is one that could easily translate to approaches valid for CNS-specific disorders.
The National Institutes of Health (NIH) has also developed a variety of programs to help investigators conduct therapeutic development programs. A few examples are outlined below.
Housed within the National Center for Advancing Translational Sciences (NCATS), the Bridging Interventional Development Gaps (BrIDGS) Program21 was originally modeled on the National Cancer Institute’s Rapid Access to Intervention Development (RAID) Program and supports preclinical research for use in an IND application with the FDA. Academic, nonprofit institutions and small businesses are eligible for this program. The research conducted is customized to the project, using contract resources and industry-trained consultants. Therapeutic modalities supported in this program range from small molecules to biologics, gene therapies, and monoclonal antibodies.
The NIH Blueprint Neurotherapeutics Network Program (BPN)22 was developed under the NIH Blueprint for Neuroscience Research, in which 10 institutes pooled their funding to support a larger scale effort in CNS drug development than what would have been feasible by an individual institution. The goal of the program is to derisk potential therapeutics to attract industry investment. The approach uses a virtual pharma model—a research investigator receives funding to support preclinical bioactivity/efficacy studies and no-cost access to contracted drug discovery and development services and consultants. Each milestone-driven drug development project accepted by the program is managed by a Lead Development Team comprised of the primary investigator (PI), seasoned industry consultants, and NIH staff. Only successful projects advance for further development, and up to $10 million is awarded to a project that meets all milestones. Due to the success of BPN for small molecules, the NIH Blueprint for Neuroscience Research recently launched the BPN for Biologics, which includes biologics-based therapies (e.g., peptides, proteins), gene-based therapies (e.g., oligonucleotide and viral-based cell therapies) and other novel emerging therapies (e.g., microbial and microbiome therapies).
Gene therapies have their own unique challenges; the NIH has begun developing resources unique to this therapeutic modality. The Foundation for NIH (FNIH) has formed the Bespoke Gene Therapy Consortium23 as a public-private partnership, including the NIH, FDA, private sector, and academic and advocacy organizations, to establish a platform for the development of gene therapies to treat rare genetic diseases. Focusing on adeno-associated virus as the gene delivery technology, the consortium aims to optimize the production, manufacturing, delivery, and target gene expression of vectors for human gene therapy using standardized trial designs and streamlined regulatory requirements.
NCATS has also launched the Platform Vector Gene Therapy (PaVe-GT) pilot program, in collaboration with National Institutes of Neurological Disorders and Stroke (NINDS) and the National Human Genome Research Institute (NHGRI), to test whether gene therapy trial start-up efficiency could be improved, using a standard approach (a single vector, single manufacturing process, and one team of researchers) for four different rare diseases: propionic acidemia, isolated methylmalonic acidemia, and two congenital myasthenic syndromes.24 All standardized templates, protocols, methods, and communications with the FDA will become publicly available for use by other researchers pursuing the development of new gene therapies.
Overall, there are a variety of available and emerging resources that can provide support for the treatment development of rare diseases. These resources have been developed by foundations and the federal government, in collaboration with clinical research organizations (CROs), industry consultants, and the FDA (where appropriate) to bring drug and gene therapy capabilities and know-how to organizations beyond the large pharmaceutical company sector.15 However, financial constraints, even in the wake of regulatory incentives, can become an impediment to drug development for orphan indications. It is worth considering that there is often significant financial pressure on patients and their families, and they often cannot access even the simple care they need. Therefore, traveling to clinical trials can be a hardship. To cover this specific need to participate in independently sponsored research, advocacy groups frequently participate in fundraising activities to help patients with day-to-day expenses and facilitate access to clinical trials. Some advocacy groups have taken more direct pathways to facilitate drug development by prioritizing investments with industry, as exemplified by the Cystic Fibrosis Foundation, where collaboration with Vertex paved the wave for a landmark approval for a novel therapeutic.25
Some people view a 501(c)3 investment into for-profit industry as a potential conflict of interest. In contrast, 502(c)3 charters generally do not allow for this type of collaboration. Because of their success, this form of collaboration will likely increase in the future.
Diversity and inclusivity: lack of available patients and researchers in key regions around the world. Successful clinical trials require that sufficient participants be available to robustly power studies to test clinically meaningful outcomes. Identifying potential study participants, as well as having a pipeline of researchers with adequate expertise to become trial investigators, is difficult for rare diseases. In some cases, well-identified centers of excellence for a particular disease already exist. If absent, therapeutic development often flounders, despite available potential drug targets. This can be overcome by proper dissemination of clinical trial information and patient-centric designs at accessible locations. However, many patients are confined to their homes or facilities and do not have access to clinical trials, and sometimes they must travel by airplane to access clinical care and participate in clinical trials.
The COVID-19 pandemic facilitated the development of decentralized trials that have enabled increased patient access to rare disease clinical trials. However, due to the complexity of digital data collection (including technical support and the restrictions of healthcare privacy laws), challenges to ensuring high quality data and delivering safe and high-quality trials remain. Ethical considerations related to conducting decentralized trials in vulnerable populations are an ongoing consideration. Risks to health and safety and data integrity versus the benefits of increased access to trials must be weighed.26
Just as there is a need to increase the number of trial participants, there also a need to increase the pipeline of clinical and translational clinical researchers working on rare disease phenomenology and treatment. Across many scientific areas, clinician scientists in research and development are decreasing in numbers; research is difficult, time-consuming, expensive, and, at times, at odds with the clinical care model of brief, infrequent patient visits.27 Flexible training, research opportunities, and a longitudinal commitment to sustaining the careers of highly trained clinician scientists are needed.28 More mechanisms to support facilities where interventional research can be conducted should be developed. It is known that study sites receive extensive training by the pharmaceutical industry in certain aspects of conduct of clinical trials; this allows PIs or co-investigators, with appropriate support, to better engage in new trials testing novel therapeutic approaches. The field can look for opportunities for private-public partnerships to establish and grow clinical centers capable of conducting drug trials in rare diseases across many countries.
Need to establish an ecosystem for drug development in rare disease. Since the advent of the Orphan Drug Act, drug development for orphan conditions has become increasingly global. With an increasing ease and ability of researchers and sponsors to recruit patients, more global drug development programs are emerging. To promote this development, the following items need to be carefully considered and implemented:
- Identification of potential differences in patient populations across cultures to facilitate inclusion of diverse populations.
- Knowledge and understanding of global regulatory requirements.
- Skill of teams to adapt the trial to improve participant experience across cultures (i.e., patient experience coordinator).
- Aggregation of rare disorders into clusters of similar disorders to leverage trial networks and trial expertise.
- Detailed sharing of successes and failure experiences for rare disease drug development, with detailed examples in the format of case studies.
Disease area ecosystems need to include patients, families, researchers, methodologists, industry/sponsors, advocacy groups, regulatory authorities, and healthcare systems whether commercial or, in some countries, nongovernmental organizations.
Development of meaningful surrogate endpoints versus biological endpoints. One of the greatest impediments to drug discovery for orphan indications is the establishment of clinically meaningful endpoints upon which regulatory agencies can approve drugs, when no biological endpoints are available or not accepted as primary endpoints by regulatory agencies. The tension between the needs of the patients and real-world outcomes must be harmonized to address the requirements of patients, caregivers, advocacy groups, regulatory agencies, public health policymakers, and researchers without losing signal detection capacity at early stages of drug development. Issues to be addressed include:
- Real-world outcomes versus agency-preferred outcomes (i.e., “primary” outcomes that are pre-established and validated by the field).
- Development of meaningful global outcome measures and endpoints that are acceptable across a variety of global cultures.
- Designs that minimize the use of placebo yet continue to allow rigorous examination of outcomes.
A thorough review of novel endpoint development and validation in rare diseases available in Busner et al.29
Development of drug targets. Obtaining stakeholder input in determining endpoints to validate drug targets is critical. This includes identifying endpoints that are meaningful to the patient and meaningful to and observable by the treating clinician. Trials must also be designed such that they can be feasibly executed and readily interpreted in patients’ lives. To accomplish these goals, the input of multiple stakeholders should be obtained. The stakeholder group should include experts on the disease state, advocates, patients, caregivers, informal caregivers, payers, and those experienced with similar trials. A feasible starting point is disease-specific qualitative research as a method to source concepts of interest that are common to several stakeholder groups. When opinions are at odds between stakeholders and cannot be fully reconciled, multiple endpoints important to the different stakeholders should be incorporated in the trial design to meet shared goals without overburdening the trial. Input should be sought early on and throughout the course of the product development process.
All stakeholders share the goal of developing effective treatments that will at best cure the disease of interest, or if not, will slow disease progression, mitigate symptoms, positively affect quality of life, and pose an acceptable risk-benefit profile, including acceptable tolerability. Obtaining stakeholder input can help in the design of a development program that avoids potential pitfalls, is time- and resource-efficient, and produces an end-product of high value because it addresses a true and acknowledged unmet need. It is also important for all stakeholders to be aware that early research efforts in rare diseases might not necessarily be aligned with a treatment or indication development process. Patients or investigators might initiate research and not have the needed experience in developing an indication or filing for approval for medications/treatments. The pathways for bringing a therapeutic to a patient can be complex, circuitous, and slow. Successful multistakeholder engagement can help address knowledge gaps, provide clarity when expectations misalign, and encourage collaborative problem-solving toward common goals.
Several initiatives, such as COSMIN,30 also promote the identification of core outcome sets specific to a disease through consensus panels involving different stakeholders to validate the most efficient measurement instruments and promote their use throughout all future clinical trials to ensure comparability. Data sharing of completed/failed clinical trials in this context could be a solution to modeling and lead to the extraction of more efficient endpoints, thus allowing for their validation in the studied patient samples before being used in a real clinical trial.
Conduct of clinical trials. Conduct of clinical trials in orphan populations requires balancing many sometimes-competing operational considerations. These include:
- Accessing a limited group of vulnerable patients.
- Collecting high quality data in areas with digital breaches.
- Issues of consent/assent, given a limited and often highly vulnerable population and uncertainty about effectiveness and safety of treatment(s).
- Limited alternative treatment options for rare diseases might overincentivize volunteerism.
- Severity of disorder and lack of known treatments might raise expectations, hope, and placebo response to a level that obscures true drug effects.
- Lack of alternatives if the experimental treatment fails, which in turn demotivates very ill candidates to take part in the studies.
- Ethical complexity to justify placebo arm, in some countries, in life-threatening early diseases.
- Common understanding or alignment with the regulatory authorities, IRBs, and health technology assessment agencies of key aspects of the study design.
When patient experience really matters. Patient experience and expertise might come from their having participated in prior research studies, while most researchers (both clinical and nonclinical) might have never taken part as a subject in any research. Study design should be carefully considered with the participants’ perspectives in mind, both via observation and obtaining direct input from participants (Table 3).

Developing treatments (medicines, devices, vaccines, diagnostics, etc.) represents a team challenge without a single, all-encompassing expert. However, assuming to have expertise in an area that is not one’s own domain when other experts are ready and willing to contribute is not a sensible step forward.
Decentralization of clinical trials (i.e., visits are conducted remotely near or at the patient’s home) might address some of the challenges noted above. Implementation of decentralized or nonlocal, site-specific trials could greatly facilitate clinical trials of orphan diseases. Less frequent site visits might be more ecological to the patient’s natural environment and could ease barriers to participation due to travel restrictions or condition-specific issues that preclude regular visits to a clinical trial site. Fewer, briefer, and less burdensome visits could also potentially minimize placebo response for certain symptoms that threaten the validity of the outcome measurement.
Although barriers to site access might impair study participation in a site-based trial, home-based visits might also adversely influence data collection and results. Previous validation of alternative evaluation formats is important prior to implementation in a study. The following list represents the general categories of considerations, with respect to home versus other remote data collection in clinical trials:
- Trial mechanics/processes are easier at site.
- Proper diagnosis of the participant’s condition is more accurate at site.
- Lack of normal routine and other interventions that could alter clinical presentation favor remote format.
- Effects of family/caregiver presence increasing patient confidence to reach the best performance possible and minimizing treatment-placebo differences favor remote format.
- Alteration of presenting symptoms due to home versus site-based clinical environment, which needs further research for each disease.
- Difficulties providing/delivering study drug to remote locations.
- Certain types of safety assessment might be more difficult to do remotely.
- Reliable collection of valid data might be challenging due to home internet issues/access.
- Monitoring of data that has been collected might not be possible in a home setting, resulting in data loss/quality concerns.
- Sponsor might require validation of “home” methodology against a site-based standard with assurance of data quality.
Hybrid study approaches should also be considered, as they could, for instance, allow different formats of the same evaluation in the same patient during the course of the study. Consideration for extent of validation required prior to alternate assessment approaches should be made on a case-by-case basis, documenting the extent of measurement equivalence.
Develop forums to convene stakeholders. Obtaining input from patients, caregivers, and advocates will increase the likelihood of identifying more patient-centered health outcomes into clinical research.
Several pre-existing forums of experts and regulators have been established, such as professional society working groups as task forces (e.g., within the International Society for CNS Clinical Trials and Methodology [ISCTM]) and government- and advocacy-funded forums. However, there is a need for more creative structures to convene stakeholders early in the disease identification and drug development process. One might consider establishing a Community Engagement Studio (Vanderbilt University, Victoria Villalta-Gil). These are moderator-led panels designed to facilitate discussion with stakeholders from underserved populations. Another mechanism might be to host community advisory boards, with a standing group of community members representing the full spectrum of affected individuals and caregivers within a certain disease stage.
A third option would be to host patient-focused drug development meetings, such as those hosted periodically by the FDA. Careful preparation and planning are required, involving a diversity of stakeholders, regions, and others to ensure all viewpoints are included. Since controversies around appropriate treatments and outcomes are likely to exist, such forums should include representation across the full disease spectrum, using electronic media to increase access and participation. Patients and career platforms, such as Carenity or PatientsLikeMe, can serve as initial stages to conduct quick polls to confirm/discard alternatives in trial designs. Unfortunately, though this is improving, patients with rare diseases are not always part of these panels. An ad hoc panel worldwide with patients with rare diseases and their caregivers would be a clear advance in the field.
Timing for inclusion of stakeholders in the research process. Early involvement of patients, caregivers, treating clinicians, expert researchers/opinion leaders, and advocacy groups is essential when working in orphan drug populations. Activities such as training caregivers on specific assessments and using video recording for endpoint monitoring are examples of such involvement. When there is a dearth of available epidemiological information related to the specific orphan disease, stakeholder inclusion should start in the pre-proof-of-concept stage. Stakeholder groups could be consulted in conjunction with Phase I studies (or earlier, if patient groups will be included) to provide the opportunity to identify and select endpoints of interest and identify potential screening, efficacy, and safety measurement tools. Prior to the initiation of proof-of-concept studies, it is critical to obtain feedback on various aspects of study design and outcome to ensure that important aspects of the disorder are addressed early in the study design process; this might be done through formal concept testing at study sites with patient panels (i.e., focus groups or direct stakeholder research) or mediated though advocacy organizations that have access to the full breadth of the study population. Design input should continue post-proof-of-concept to assess whether the results obtained and the risk-benefit ratio are favorable and identify what changes should be made to improve the research process during confirmatory trials. It might also be useful for sponsors to engage with the broad community about how to best position risk versus benefit and in preparation for approval and launch. Regional and cultural considerations are important to identify at each phase of research, as communities might be isolated from one another, or awareness and diagnosis might be regionally variable.
Some recommendations include the following:
- Involve diverse community members as advisors to the protocol team.
- Pretest protocol procedures to confirm feasibility with patient panels.
- When involving caregivers, parents, and advocates, scientists must use lay language and assure understanding of protocol/treatment goals.
- Consider groups, such as ISCTM Working Groups, to serve as a host for standalone meetings or teleconferences to assist in the development of research strategies
- Work with NORD and other groups
- Involve the National Institute of Mental Health (NIMH) and National Institute of Child Health and Human Development (NICHD) as advisory resources for the following:
- Natural history protocols, study design, development of trial sites, meaningful endpoints, and feasibility
- A source of opinions on meaningful outcomes
- A source of trials subjects
- Data access agreements for future uses
Other special considerations. As environments change, the need to design trials and access patients is affected. For example, in countries that are plagued by war, access to patients might be limited or prohibited, or patients may be impossible to retain. In other locales, patient access could be limited by mores, fear of experimentation, or other local conditions or cultural beliefs. Also, governmental health systems in some countries might constitute a barrier to identifying health establishments that attend to patients with rare diseases. Back-up plans for other issues, such as natural disasters or other unforeseen conditions that could affect clinical trials, might need to be developed.
In the case of the COVID-19 pandemic, decentralized clinical trials undertaken at home might impact outcome measures, such as those potentially seen in autism clinical trials. For example, under the current COVID-19 conditions, the shelter-in-place orders that required home confinement might not be the best set of conditions under which to access behavior outcomes. Aman and Pearson31 outlined some of the challenges of data collection for children with autism during the COVID-19 pandemic and the “moving target” type of data collection that could be done under various conditions. While some subjects in a trial could have had all of their data collected pre-COVID-19 (A), others could have had it collected both before COVID-19 and during COVID-19 (B), both during COVID-19 and after COVID-19 (C), and after COVID-19 (D) (Figure 1).
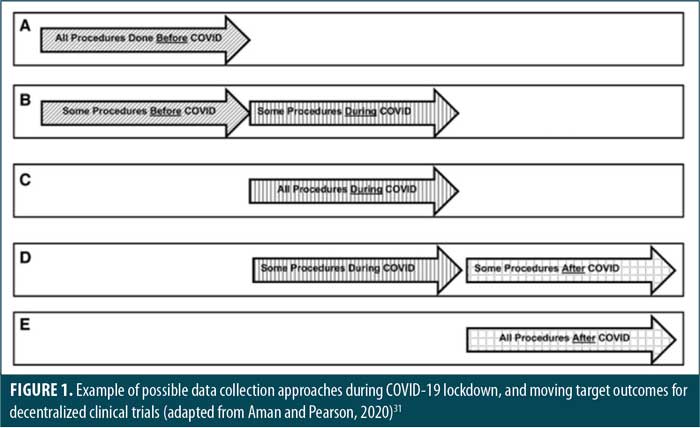
The assurance of comparable data quality of remote and in-person assessments and the means of evaluating the additional degrees of freedom introduced with alternate modalities are among the many challenges to be addressed. The authors also noted the difficulties in assessing the generalizability of moving target data; comparisons and other data analysis would be quite difficult with moving target data collection as well. To date, questions remain as to how the various regulatory agencies view this data and whether collecting data under these circumstances will allow for the proper power and other statistical considerations for a valid, well-controlled trial.
Busner32 also identified some of the COVID-19 pandemic-related challenges to be considered in the design and interpretation of pediatric trials.
Conclusion
Rare diseases represent an area of significant unmet medical need. Involving the full range of stakeholders can facilitate improved study designs, as well as better conduct and more efficient clinical outcomes for individuals affected by rare disease. The development of global specific platforms to facilitate information exchange along the research process is needed; this should include drug developers, patients, caregivers, clinicians, experts, funders, and others. The implementation of existing best practices and continued shaping of the ecosystem surrounding rare diseases are important opportunities for advancing the field. Providing facilities to independent researchers in planning and conducting clinical trials in healthcare settings is also a potentially promising path. Private-public initiatives that include patients and caregivers worldwide are needed and will continue to raise public understanding and interest and boost high quality clinical trials of new interventional strategies in rare diseases.
References
- Nguengang Wakap S, Lambert DM, Olry A, et al. Estimating cumulative point prevalence of rare diseases: analysis of the Orphanet database. Eur J Hum Genet. 2020;28(2):165–173.
- Mazzucato M., Visonà Dalla Pozza L, Minichiello C, et al. Estimating mortality in rare diseases using a population-based registry, 2002 through 2019. Orphanet J Rare Dis. 2023;18(1):362.
- Groft SC, Posada M, Taruscio D. Progress, challenges and global approaches to rare diseases. Acta Paediatr. 2021;110(10):2711–2716.
- Mikami K, Sturdy S. Patient organization involvement and the challenge of securing access to treatments for rare diseases: report of a policy engagement workshop. Res Involv Engagem. 2017;3:14.
- Chan AYL, Chan VKY, Olsson S, et al. Access and unmet needs of orphan drugs in 194 countries and 6 areas: a global policy review with content analysis. Value Health. 2020;23(12):1580–1591.
- Mireku A. Can the FDA keep the momentum going for rare disease drug approvals? Pharmaceutical Technology. 23 Jan 2023. https://www.pharmaceutical-technology.com/features/can-the-fda-keep-the-momentum-going-for-rare-disease-drug-approvals/. Accessed 10 Jan 2024.
- Dunkle M, Pines W, Saltonstall PL. Advocacy groups and their role in rare diseases research. Adv Exp Med Biol. 2010;686:515–525.
- National Organization for Rare Disorders. https://rarediseases.org/. Accessed 10 Jan 2024.
- National Organization for Rare Disorders. Global partnerships. https://rarediseases.org/community-support/global-partnerships/. Accessed 10 Jan 2024.
- International Rare Diseases Research Consortium. https://irdirc.org/. Accessed 10 Jan 2024.
- Baird DA, Liu JZ, Zheng J, et al. Identifying drug targets for neurological and psychiatric disease via genetics and the brain transcriptome. PLoS Genet. 2021;17(1):e1009224.
- Condò I. Rare monogenic diseases: molecular pathophysiology and novel therapies. Int J Mol Sci. 2022;23(12):6525.
- Song J, Bang S, Choi N, Kim HN. Brain organoid-on-a-chip: a next-generation human brain avatar for recapitulating human brain physiology and pathology. Biomicrofluidics. 2022;16(6):061301.
- Vatansever S, Schlessinger A, Wacker D, et al. Artificial intelligence and machine learning-aided drug discovery in central nervous system diseases: state-of-the-arts and future directions. Med Res Rev. 2021;41(3):1427–1473.
- D’Angelo CS, Hermes A, McMaster CR, et al. Barriers and considerations for diagnosing rare diseases in indigenous populations. Front Pediatr. 2020;8:579924.
- Lotan M, Downs J, Stahlhut M, Romano A. Evaluation tools developed for Rett syndrome. Diagnostics (Basel). 2023;13(10):1708.
- Lee BH, Deng S, Chiriboga CA, et al. Newborn screening for spinal muscular atrophy in new York state: clinical outcomes from the first 3 years. Neurology. 2022;99(14):e1527–1537.
- Cook NS, Cave J, Holtorf AP. Patient preference studies during early drug development: aligning stakeholders to ensure development plans meet patient needs. Front Med (Lausanne). 2019;6:82.
- Yu D. An overview of recent US-approved gene therapies for Duchenne muscular dystrophy and their respective clinical development programs. Drugs Ther Perspect. 2023;39:156–163.
- Ramsey BW, Davies J, McElvaney NG, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med. 2011;365(18):1663–1672.
- National Center for Advancing Translationals Sciences. BrIDGs. https://ncats.nih.gov/research/research-activities/bridgs. Accessed 16 Feb 2024.
- National Instititutes of Health. The NIH Blueprint for Neuroscience Research. https://neuroscienceblueprint.nih.gov/. Accessed 16 Feb 2024.
- Foundation for the National Institutes of Health. AMP® Bespoke Gene Therapy Consortium (BGTC). https://fnih.org/BGTC. Accessed 10 Jan 2024.
- Brooks PJ, Ottinger EA, Portero D, et al. The Platform Vector Gene Therapies project: increasing the efficiency of adeno-associated virus gene therapy clinical trial startup. Hum Gene Ther. 2020;31(19–20):1034–1042.
- Cystic Fibrosis Foundation. Our venture philanthropy model. https://www.cff.org/about-us/our-venture-philanthropy-model. Accessed 10 Jan 2023.
- Petrini C, Mannelli C, Riva L, et al. Decentralized clinical trials (DCTs): a few ethical considerations. Front Public Health. 2022;10:1081150.
- Weggemans MM, van der Schaaf M, Kluijtmans M, et al. Preventing translational scientists from extinction: the long-term impact of a personalized training program in translational medicine on the careers of translational scientists. Front Med (Lausanne). 2018;5:298.
- Noble K, Owens J, André F, et al. Securing the future of the clinician-scientist. Nat Cancer. 2020;1(2):139–141.
- Busner J, Pandina G, Domingo S, et al. Clinician- and patient-reported endpoints in CNS orphan drug clinical trials: ISCTM position paper on best practices for endpoint selection, validation, training, and standardization. Innov Clin Neurosci. 2021;18(10–12):15–22.
- COSMIN. https://www.cosmin.nl/. Accessed 10 Jan 2024.
- Aman MG, Pearson DA. Challenges for child and adolescent psychiatric research in the era of COVID-19. J Child Adolesc Psychopharmacol. 2020;30(5)280–284.
- Busner J. Special considerations for child psychiatric clinical trials during a global pandemic. J Clin Stud. 2020;12(4):46–48.