

by Muhannad M. Alsharidah, MBBS; Mohammad Uzair, BS; Sarah S. Alseneidi, MBBS; Afnan A. Alkharan, MBBS;
Reem Fahd Bunyan, MBBS; and Shahid Bashir, PhD
Drs. Alsharidah and Alseneidi are with the College of Medicine, King Saud University in Riyadh, Saudi Arabia. Mr. Uzair is with the Department of Biological Sciences, Faculty of Basic and Applied Sciences, International Islamic University in Islamabad, Pakistan. Dr. Alkharan is with the College of Medicine, Princess Nourah Bint Abdulrahman University in Riyadh, Saudi Arabia. Drs. Bunyan and Bashir are with the Neuroscience Center, King Fahad Specialist Hospital in Dammam, Saudi Arabia.
Funding: No funding was provided for this study.
Disclosures: The authors have no conflicts of interest relevant to the content of this article.
Innov Clin Neurosci. 2022;19(1–3):8–14.
ABSTRACT: Objective: Multiple sclerosis (MS) is a chronic, immune-mediated inflammatory disease of the central nervous system (CNS) characterized by demyelination, axonal degeneration, and cognitive impairment. It also has an important impact on the quality of life of patients and their family members. We sought to determine if transcranial magnetic stimulation (TMS) is a valid instrument for determining disease progression activity in patients with MS.
Methods: A literature search of the PubMed database was conducted using the terms “multiple sclerosis,” “transcranial magnetic stimulation,” and “neurophysiological parameters.”
Results: Neurophysiological parameters, such as sensitivity to demyelination and the strength of excitatory and inhibitory synaptic interactions in the cerebral cortex, can be identified through TMS in patients affected by MS. These objective parameters can be correlated with the progression of disease and provide reliable indices for the severity of illness and the efficacy of drugs used to treat MS in clinical trials.
Conclusion: The discovery of specific and detailed neurophysiological parameters as surrogate endpoints for disease activity could represent an important step in clinical trials. Changes in cortical connectivity have already been demonstrated in MS, but in clinical practice, other measures are typically used to evaluate disease activity. We speculate that TMS might be more effective in identifying disease progression that leads to long-term disability, compared to standard surrogate markers, since it represents a direct measure of synaptic transmission(s) in MS.
Keywords: Multiple sclerosis, transcranial magnetic stimulation, evoked potentials
Multiple sclerosis (MS) is a chronic, immune-mediated inflammatory disease of the central nervous system (CNS) characterized by demyelination, axonal degeneration, and cognitive impairment. It also has an important impact on the quality of life of patients and their family members.1
MS is the leading cause of nontraumatic neurological disabilities in young adults, affecting 0.1 percent of the general population in Western countries.2,3 Several innovative new drugs are currently in advanced stages of development for MS. The principal clinical measures of disease activity used to assess the efficacy of new drugs for MS in clinical trials, such as the Expanded Disability Status Scale and CNS magnetic resonance imaging (MRI), present some limitations.4 Numerous studies have investigated changes in cortical excitability using transcranial magnetic stimulation (TMS) in MS as a tool to determine disease severity.5,6 TMS could represent a valid instrument for obtaining objective and reliable markers and potential surrogate endpoints for the progression of MS.
TMS, a safe, inexpensive, and noninvasive neurophysiological technique, detects abnormalities in patients with MS that can be correlated with the severity of clinical manifestation, as well as the brain and spinal cord MRI lesion load.7,8 If validated, TMS could be used as a surrogate marker of disease activity and represent a suitable measure of drug efficacy in MS clinical trials. The use of neurophysiological techniques in MS clinical trials, such as TMS, could shorten their duration, reduce their costs (compared to MRIs), allow greater accessibility to the majority of clinical institutions, and provide more reliable data on drug efficacy.9 Thus, TMS could contribute to the development of improved treatments for the millions of individuals currently affected by MS around the world. We sought to determine the validity of TMS as a tool for determining disease severity and drug efficacy of MS in clinical trials.
Historical Context
Jean-Martin Charcot10 is generally credited as the first person to comprehensively characterize MS as a distinct disease. During a series of lectures entitled “Les scleroses en plaques disseminate,” presented in the Salpetriere Hospital in Paris, Charcot described a condition occurring in younger adults who, at autopsy, were noted to have grayish and reddish plaques of variable contours and sizes scattered through the CNS. Charcot gave an account of their clinical features, delineating the cerebral, spinal, and mixed cerebrospinal structures with vivid descriptions of the clinical pathogenesis and pathophysiology. He identified the discrepancy between lesions and symptoms and established the link between axonal loss and clinical disability.11
Prevalence and incidence. MS most commonly affects young adults (average age: 28 years), with predominance among female individuals.2,12 However, the association between aging and the clinical course of MS is still elusive. According to the Atlas of MS, approximately 2.8 million people (35.9 per 100,000) suffer from MS worldwide, 67 percent of whom are women.13 The United States (US) reported the highest number of MS cases, with about 913,925 (288 per 100,000 population) in 2021, and Germany has the highest prevalence rate at 303 per 100,000 people (Figures 1A and 1B). Death is attributed to MS in two-thirds of cases. The high national prevalence rate and the long-term nature of the disease place a significant financial burden on the economy of the Kingdom of Saudi Arabia (KSA).14,15 The projected prevalence of MS in Saudi Arabia was reported to be 40.40/100,000 total population with a higher prevalence among Saudi-nationals (61.95/100,000).16
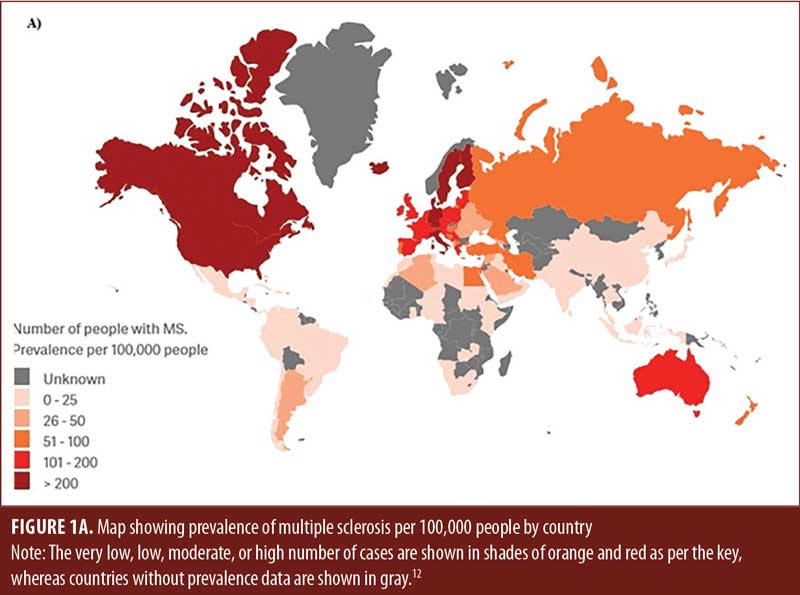
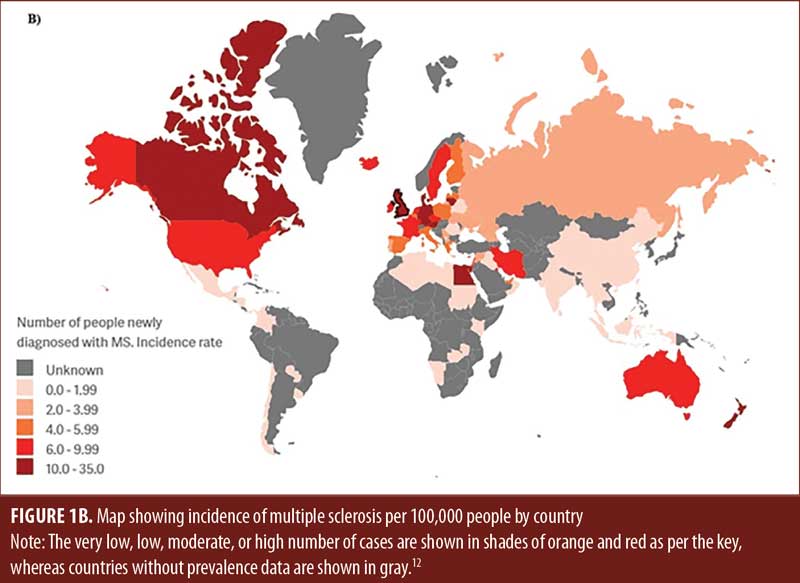
Diagnosis. For most people, the clinical course of MS begins with episodes of neurological dysfunction, followed by chronic remitting-relapsing episodes.17 MRI is the gold standard for diagnosing MS and monitoring the course of the disease.18,19 However, although MRI scans can reveal multiple lesions distributed throughout the white and gray matter of the CNS,20 diagnosis of MS with MRI is frequently confounded by the differential diagnosis of other diseases characterized by demyelination of the optic nerves (neuritis), severe myelopathy with extensive spinal cord lesions, or even a normal MRI with abnormalities typical to MS.21 Therefore, cerebrospinal fluid (CSF) analysis is often employed to confirm the diagnosis. A persistent and consistent presence of oligoclonal banding (OCB) exists in the CSF of patients with MS; thus, a positive CRF analysis requires only two lesions, identified by MRI, to confirm the diagnosis of MS.22,23
Lesions typically affect the optic nerve, brainstem, cerebellum, spinal cord, and/or cerebral hemispheres.24 However, if white matter abnormalities are detected by MRI at clinically unaffected sites, the probability of a future positive diagnosis increases from 50 percent within two years to 82 percent within 20 years.24
Classification of MS. A confirmed diagnosis of MS requires evidence of characteristic neurological lesions in the CNS that have occurred in both time and space, as well as exclusion of alternative diagnoses.18
Etiology. Epidemiological studies of MS indicate a complex etiology in which unidentified environmental factors trigger the disease in genetically susceptible individuals.25 MS is linked to alleles of the major histocompatibility complex (MHC). Although the exact mechanisms are unknown, the human leukocyte antigen HLA-DRB1*1501 gene is the strongest genetic factor identified to date that influences susceptibility to MS.26 This gene might also determine the balance between disease susceptibility and resistance.25,26
MS has a familial recurrence rate of about 17.3 percent.27 When both parents have MS, the risk increases to about 30 percent.27 However, thus far, investigations into the recurrence of familial MS and studies relating to monozygotic and dizygotic twins have provided no conclusive evidence of the presence of hereditary genetic traits.28
Demyelination. Myelin is formed from the extending plasma membrane of oligodendrocytes, creating spiral segments of sheathing that wrap around axons and envelop bundles of axonal segments. The insulating and protective properties of the myelin sheath are largely due to its structure, thickness, low water content, and richness in lipids. Myelin sheath thicknesses and internodal lengths vary according to axonal caliber.29 The number of wrappings around an axon can vary between 10 and 160.30 Depending on the subtype of the oligodendrocyte, 10 or more axons can be myelinated at one time.31 As a consequence, inflammatory damage to a single oligodendrocyte has the potential to affect multiple axons.
Remyelination. Remyelination is associated with functional recovery in MS, although some individuals have demonstrated extensive remyelination without signs of functional improvement.32 However, the extent of remyelination between individuals, or even within specific MS lesions, is highly variable.33 Thus, even in the progressive forms of MS, compensatory mechanisms have been observed that respond to inflammatory injury in the neural circuits. In some cases, this allows for partial or complete restoration of neural function.32 However, there are many disturbances to which the CNS cannot respond and where the insult and injury could be of such intensity that any natural response is inadequate.34
Cellular abnormalities. Traditionally, axonal degeneration has been regarded as the major cause of neurological deficits and irreversible disability in pulse-width modulation.35 However, while it is acknowledged that the symptoms of MS are generally attributable to the interruption of myelinated tracts in the CNS,36 recent studies have shown that a certain proportion of neurodegeneration is independent of demyelination.3,33 Indeed, the current view is that the entire CNS appears to be involved in the disease.37 Inflammatory processes within the CNS can trigger a cascade of events that profoundly affect synaptic density, neurotransmitter concentrations, signal transmission mechanisms, and mitochondrial density and disrupt the microtubule transport systems critical to the normal functioning and survival of neurons.38 The extent of MS-related symptoms and disability is determined by the intrinsic ability of the CNS to retain the integrity and compliance of the central mechanisms and neural pathways.39
Glutamate. Glutamate is the principal fast excitatory neurotransmitter in the CNS. Glutamate-dependent signaling is required for all sensory and motor processing, and glutamatergic receptors contribute significantly to synaptic plasticity, as well as learning and memory.40 During inflammatory episodes, microglia and leukocytes release substantial amounts of glutamate into the extracellular space.41 N-methyl-D-aspartate (NMDA) receptors consist of a complex pharmacology with multiple modulatory sites. They are key components in long-term potentiation (LTP) of neuronal pathways, memory formation, and synaptic plasticity.42 Α‐amino‐3‐hydroxy‐5‐methylisoxazole‐4‐propionic acid (AMPA) receptors desensitize quickly, strongly influencing neuronal output, and are responsible for the majority of fast excitatory transmissions and enhanced synaptic plasticity in the human brain.43 Recently, confocal microscopy has revealed that MS causes a significant decrease in excitatory synapses and mRNA proteins encoding AMPA, NMDA, and metabotropic glutamate (mGlu) receptors.44 Moreover, a significant reduction in their respective synaptic binding proteins within the hippocampi of post-mortem MS brains has been reported.44,45
Gama-aminobutyric acid (GABA). Gama-aminobutyric acid (GABA) receptors and glycine are the principal inhibitory transmitters in the CNS that, when activated, can generate membrane hyperpolarization and reduction in dendritic excitatory activity, which strongly inhibits action potential firing.46 GABA, as well as GABAergic receptors, have been found to be significantly elevated in MS demyelinated hippocampi, suggesting that GABA exerts a powerful inhibitory influence in MS.47
Medication. Disease-modifying therapies for MS are still at a relatively early stage of development. Drugs have been shown to be effective in reducing relapse rates in the early forms of the disease, with a 30-percent reduction in frequency of new episodes over 2 to 3 years, and, to a certain extent, slowing clinical progression during relapsing-remitting episodes.48 However, studies have shown no useful effects of drug therapies on the secondary-progressive phase, except in rare cases of progressive forms of the disease with continuing high relapse rates.49 Long-term observational studies assessing the validity of drug interventions have suffered from the lack of patient willingness to participate in the studies or, in some cases, loss to follow-up or death.50 Initially, there appears to be a high level of adherence to medications, but discontinuation rates have been reported as high as 50 percent within two years of drug initiation.50
Summary. MS presents as a complex, unpredictable, changeable, and heterogeneous disease affecting the CNS, causing moderate-to-severe disability in the majority of those affected by it. Symptoms can change without warning, leading to an unpredictable level of dysfunction and recovery.51 The fluctuating and progressive nature of this disease can present a confusing picture to clinicians, complicating its management.52
Neurophysiological Measures
Transcranial magnetic stimulation. TMS is a well-established, noninvasive technique used to examine the integrity and excitability of the corticospinal-neuromuscular pathway in both healthy and neurologically impaired populations.53 The following sections describe the technical principles underlying TMS and the methodological considerations and safety issues that influence the design of TMS experiments.7 A single stimulus evokes multiple descending volleys in corticospinal motor neurons, producing a contralateral, synchronous muscle response, also known as a motor-evoked potential (MEP).54 Surface electromyography (EMG) electrodes are placed on the target muscles to be stimulated, and, once positioned, the magnetic coil is fired and MEPs are recorded by surface EMG electrodes.8 The repetitive discharge of spinal motor neurons represents the natural excitability of the corticospinal-neuromuscular muscular pathway in response to the magnetic pulses of the TMS.55
MEP variability. The MEP is influenced by intrinsic factors relating to the excitability of the corticospinal pathway, such as the number of motor neurons recruited by the magnetic pulse, the number of motor neurons discharging more than once in response to the stimulus, and the synchronization of the motor neuronal discharges.56,57 The stimulus-response relationship varies considerably between subjects and between muscles in the same individual, even those in close anatomical proximity, such as hand muscles or upper leg muscles.56 Extrinsic factors that are implicated in MEP variability are the role of the coil and its position on the scalp. Furthermore, MS-related conduction block, conduction impedance, and slowing of signal velocity might modify the MEP.58
TMS measures and motor threshold. The TMS measures most commonly used in the analysis of motor data are resting motor threshold (MT; the minimal TMS intensity required to evoke an MEP), latency (the time from triggering the pulse to the start of the MEP), amplitude (baseline to peak or peak to peak value), and area (measured as root mean squared or as the area under the line of the rectified signal).59 MT is the lowest stimulus intensity of TMS that elicits a recordable MEP in the target muscle.60 Obtaining accurate resting MTs requires a systematic search for the optimal coil position (i.e., hotspot) above the motor area corresponding to the target limb or muscle.61 When the coil is directly over the hotspot, it is a general practice to obtain 5 to 10 MEPs of 50µV in at least 50 percent of successive trials.62 MTs are generally recognized as a measure of the integrity and excitability of the neural pathway, at both the cortical and spinal level.63
Central motor conduction time. The latency of an evoked potential is the time interval between triggering the stimulus at the motor cortex and the start of the MEP.64 The time required for a volley generated in the motor cortex to travel through the spinal cord is known as the central motor conduction time.65 It can be determined by comparing the difference between the latency of a cortically stimulated MEP and the latency of a TMS pulse at the spinal roots or the latency of the response to electrical or magnetic stimulation at a point on the motor nerve.66
The cortical silent period. The cortical silent period (CSP) is characterized by an interruption of the EMG signal after the TMS is delivered at the motor cortex during a tonic contraction.67 It is defined as the time between the end of the MEP and the return of the EMG signal.68 CSP requires an individual to contract the target muscle at about 20 to 30 percent of maximal voluntary force.69 The duration of the CSP is dependent upon the percent of stimulator output, yet it appears to be unaffected by the intensity of the maximum voluntary contraction (MVC).70 The resulting CSP lasts for about 100ms, with considerable variability in duration between subjects.71 The initial portion (40–50ms) of the CSP is generally believed to be a response to spinal inhibitory mechanisms that follow motor neuron excitation, namely hyperpolarization and recurrent inhibition. Inhibitory mechanisms at the spinal level are known to mediate the CSP during its latter stages.71
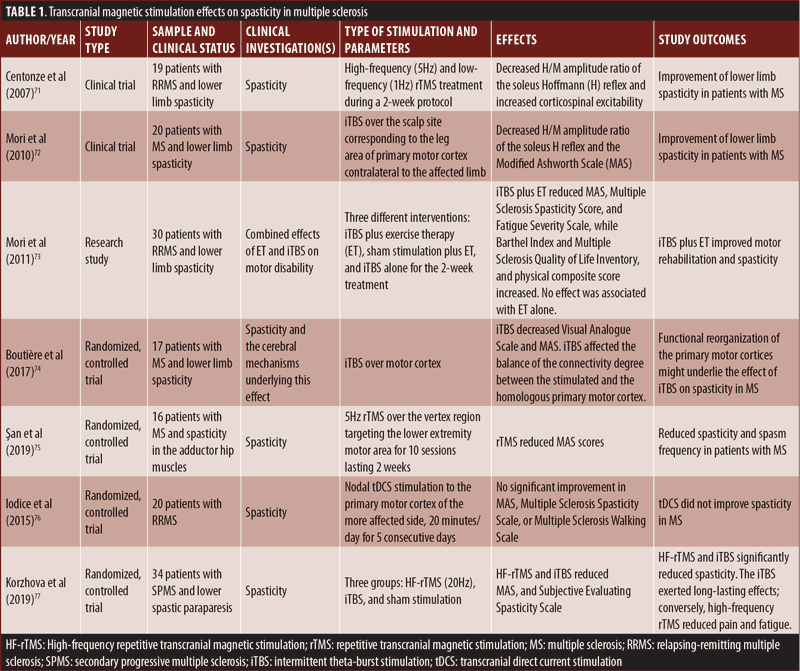
TMS effects on spasticity in MS. Spasticity affects 60 to 80 percent of patients with MS, causing gait disorders, spasms, fatigue, falls, sleep disorders, and muscle tightness, thus worsening disability.72 However, TMS could be a better choice of treatment, as it effectively treats spasticity (Table 1). Centonze et al73 applied high-frequency (5Hz) and low-frequency (1Hz) repetitive TMS (rTMS) over the primary motor cortex (M1) of the patients with relapsing-remitting MS and lower limb spasticity. High-frequency rTMS increased MEP amplitude and corticospinal excitability and improved lower limb spasticity. A single session of rTMS did not exert observable effects of spasticity. Low-frequency rTMS had the opposite outcomes. The study suggests that rTMS has significant therapeutic effects on spasticity in MS by promoting lasting modifications of complex spinal circuits.73 The intermittent theta-burst stimulation (iTBS), a form of rTMS treatment is also effective in improving lower limb spasticity in patients with MS. Studies by Mori et al74 and Mori et al75 reported that iTBS reduces spasticity and fatigue in patients with MS. iTBS induced long-term changes in cortical excitability and LTP-like changes at synaptic connections in the motor cortex, changing corticospinal synapses activity.74 Boutière et al76 evaluated whether iTBS of the M1 regions induced modulation of spasticity, which is associated with the functional reorganization of M1 regions in patients with MS. The authors observed that the iTBS treatment had significant effects on interhemispheric balance, which were correlated with improvement of spasticity.76 A recent study combined physical therapy and a rehabilitation program with 5Hz rTMS over the vortex region targeting the lower extremity motor area of the cerebral cortex. The study concluded that long-term rTMS treatment, combined with physical therapy, with a one-month follow-up reduces spasticity in patients with MS more than physical therapy alone.77 However, anodal transcranial direct current stimulation (tDCS) over M1 does not improve lower limb spasticity in patients with relapsing-remitting MS.78 A recent study compared the effects of high-frequency rTMS and iTBS on spasticity and concomitant symptoms in patients with secondary progressive MS. The study demonstrated that both high-frequency rTMS and iTBS reduced spasticity in contrast to sham stimulation. The iTBS exerted long-lasting effects, and, conversely, high-frequency rTMS reduced pain and fatigue.79 While more studies are required with a large cohort of patients with MS, noninvasive brain stimulation could be an efficient therapeutic approach for motor rehabilitation and managing spasticity in patients with MS.
Conclusion
Neurological diseases frequently show typical patterns of cortical excitability, which are likely to change during the progression of the illness.80 Our working hypothesis is that altered patterns of excitability tend to regress to normal activity under effective therapeutic approaches. The discovery of specific and detailed neurophysiological parameters as a surrogate endpoint of disease activity could represent an important step in clinical trials, providing reliable indices of severity of illness and efficacy of drugs.
Changes in cortical connectivity have already been demonstrated in MS, but in clinical practice, other measures are typically used to evaluate disease activity. We speculate that TMS might be more effective in signaling disease progression that leads to long-term disability, compared to standard surrogate markers, because it represents a direct measure of synaptic transmission(s).
Author Contributions
All authors contributed in writing and editing the draft.
References
- Homayuni A, Abedini S, Hosseini Z, et al. Explaining the facilitators of quality of life in patients with multiple sclerosis: a qualitative study. BMC Neurol. 2021;21(1):193.
- Walton C, King R, Rechtman L, et al. Rising prevalence of multiple sclerosis worldwide: insights from the Atlas of MS, third edition. Mult Scler. 2020;26(14):1816–1821.
- Alonso A, Hernán MA. Temporal trends in the incidence of multiple sclerosis: a systematic review. Neurology. 2008;71(2):129–135.
- Karabudak R, Dahdaleh M, Aljumah M, et al. Functional clinical outcomes in multiple sclerosis: current status and future prospects. Mult Scler Relat Disord. 2015;4(3):192–201.
- Ayache SS, Créange A, Farhat WH, et al. Cortical excitability changes over time in progressive multiple sclerosis. Funct Neurol. 2015;30(4):257–263.
- Neva JL, Lakhani B, Brown KE, et al. Multiple measures of corticospinal excitability are associated with clinical features of multiple sclerosis. Behav Brain Res. 2016;297:187–195.
- Simpson M, Macdonell R. The use of transcranial magnetic stimulation in diagnosis, prognostication and treatment evaluation in multiple sclerosis. Mult Scler Relat Disord. 2015;4(5):430–436.
- Hsu WY, Cheng CH, Zanto TP, et al. Effects of transcranial direct current stimulation on cognition, mood, pain, and fatigue in multiple sclerosis: a systematic review and meta-analysis. Front Neurol. 2021;12:626113.
- Houdayer E, Comi G, Leocani L. The neurophysiologist perspective into MS plasticity. Front Neurol. 2015;6:193.
- Makris A, Piperopoulos A, Karmaniolou I. Multiple sclerosis: basic knowledge and new insights in perioperative management. J Anesth. 2014;28(2):267–278.
- Przybek J, Gniatkowska I, Mirowska-Guzel D, Członkowska A. Evolution of diagnostic criteria for multiple sclerosis [published correction appears in Neurol Neurochir Pol. 2016;50(4):321.]. Neurol Neurochir Pol. 2015;49(5):313–321.
- Gomes Mda M, Engelhardt E. Jean-Martin Charcot, father of modern neurology: an homage 120 years after his death. Arq Neuropsiquiatr. 2013;71(10):815–817.
- Multiple Sclerosis International Federation. Atlas of MS, 3rd Edition. 2020. www.atlasofms.org. Accessed 18 Jan 2022.
- Grytten N, Torkildsen Ø, Myhr KM. Time trends in the incidence and prevalence of multiple sclerosis in Norway during eight decades. Acta Neurol Scand. 2015;132(199):29–36.
- Kristofikova Z, Gazova Z, Siposova K, et al. Effects of ferrofluid and phytoalexin spirobrassinin on thioflavin-T-based fluorescence in cerebrospinal fluid of the elderly and multiple sclerosis patients. Neurochem Res. 2014;39(8):1502–1510.
- AlJumah M, Bunyan R, Al Otaibi H, et al. Rising prevalence of multiple sclerosis in Saudi Arabia, a descriptive study. BMC Neurol. 2020;20(1):49.
- Daif AM. Multiple sclerosis. Recent modalities of treatment. Neurosciences (Riyadh). 2004;9(3):143–149.
- Alamri Y, Al-Busaidi IS. Multiple sclerosis in Saudi Arabia: anxiety, depression and suicidality. Psychiatry Res. 2016;238:24.
- Dutta R, Trapp BD. Relapsing and progressive forms of multiple sclerosis: insights from pathology. Curr Opin Neurol. 2014;27(3):271–278.
- Tripathi A, Pandit I, Perles A, et al. Identifying miRNAs in multiple sclerosis gray matter lesions that correlate with atrophy measures. Ann Clin Transl Neurol. 2021;8(6):1279–1291.
- Garg N, Smith TW. An update on immunopathogenesis, diagnosis, and treatment of multiple sclerosis. Brain Behav. 2015;5(9):e00362.
- Jacobsen CO, Farbu E. MRI evaluation of grey matter atrophy and disease course in multiple sclerosis: an overview of current knowledge. Acta Neurol Scand Suppl. 2014;(198):32–36.
- Wattjes MP, Rovira À, Miller D, et al. Evidence-based guidelines: MAGNIMS consensus guidelines on the use of MRI in multiple sclerosis—establishing disease prognosis and monitoring patients. Nat Rev Neurol. 2015;11(10):597–606.
- Haghikia A, Haghikia A, Hellwig K, et al. Regulated microRNAs in the CSF of patients with multiple sclerosis: a case-control study. Neurology. 2012;79(22):2166–2170.
- Stich O, Kluge J, Speck J, Rauer S. Oligoclonal restriction of antiviral immunoreaction in oligoclonal band-negative MS patients. Acta Neurol Scand. 2015;131(6):381–388.
- Chen W, Gauthier SA, Gupta A, et al. Quantitative susceptibility mapping of multiple sclerosis lesions at various ages. Radiology. 2014;271(1):183–192.
- Sawcer S. A new era in the genetic analysis of multiple sclerosis. Curr Opin Neurol. 2006;19(3):237–241.
- Link J, Kockum I, Lorentzen AR, et al. Importance of human leukocyte antigen (HLA) class I and II alleles on the risk of multiple sclerosis. PLoS One. 2012;7(5):e36779.
- Hader WJ, Yee IM. The prevalence of familial multiple sclerosis in Saskatoon, Saskatchewan. Mult Scler Int. 2014;2014:545080.
- Sadovnick AD. European Charcot Foundation Lecture: the natural history of multiple sclerosis and gender. J Neurol Sci. 2009;286(1–2):1–5.
- Lubetzki C, Stankoff B. Demyelination in multiple sclerosis. Handb Clin Neurol. 2014;122:89–99.
- Lee JY, Parisi TJ, Friedrich PF, Bet al. Does the addition of a nerve wrap to a motor nerve repair affect motor outcomes? Microsurgery. 2014;34(7):562–567.
- Lee DH, Linker RA. The role of myelin oligodendrocyte glycoprotein in autoimmune demyelination: a target for multiple sclerosis therapy? Expert Opin Ther Targets. 2012;16(5):451–462.
- Olsen JA, Akirav EM. Remyelination in multiple sclerosis: cellular mechanisms and novel therapeutic approaches. J Neurosci Res. 2015;93(5):687–696.
- Williams A. Remyelination in multiple sclerosis: what do we know and where are we going?. Neurodegener Dis Manag. 2015;5(1):49–59.
- Tanaka T, Yoshida S. Mechanisms of remyelination: recent insight from experimental models. Biomol Concepts. 2014;5(4):289–298.
- Campbell GR, Worrall JT, Mahad DJ. The central role of mitochondria in axonal degeneration in multiple sclerosis. Mult Scler. 2014;20(14):1806–1813.
- Fox RJ, Bacon TE, Chamot E, et al. Prevalence of multiple sclerosis symptoms across lifespan: data from the NARCOMS Registry [published correction appears in Neurodegener Dis Manag. 2016;6(2):178]. Neurodegener Dis Manag. 2015;5(6 Suppl):3–10.
- Kutzelnigg A, Lucchinetti CF, Stadelmann C, et al. Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain. 2005;128(Pt 11):2705–2712.
- Friese MA, Schattling B, Fugger L. Mechanisms of neurodegeneration and axonal dysfunction in multiple sclerosis. Nat Rev Neurol. 2014;10(4):225–238.
- Chamberlain KA, Nanescu SE, Psachoulia K, Huang JK. Oligodendrocyte regeneration: its significance in myelin replacement and neuroprotection in multiple sclerosis. Neuropharmacology. 2016;110(Pt B):633–643.
- Stojanovic IR, Kostic M, Ljubisavljevic S. The role of glutamate and its receptors in multiple sclerosis. J Neural Transm (Vienna). 2014;121(8):945–955.
- Kostic M, Dzopalic T, Zivanovic S, et al. IL-17 and glutamate excitotoxicity in the pathogenesis of multiple sclerosis. Scand J Immunol. 2014;79(3):181–186.
- Fleischmann R, Prüss H, Rosche B, et al. Severe cognitive impairment associated with intrathecal antibodies to the NR1 subunit of the N-methyl-D-aspartate receptor in a patient with multiple sclerosis. JAMA Neurol. 2015;72(1):96–99.
- Jansson LC, Åkerman KE. The role of glutamate and its receptors in the proliferation, migration, differentiation and survival of neural progenitor cells. J Neural Transm (Vienna). 2014;121(8):819–836.
- Azevedo CJ, Kornak J, Chu P, et al. In vivo evidence of glutamate toxicity in multiple sclerosis. Ann Neurol. 2014;76(2):269–278.
- Sulkowski G, Dąbrowska-Bouta B, Salińska E, Strużyńska L. Modulation of glutamate transport and receptor binding by glutamate receptor antagonists in EAE rat brain. PLoS One. 2014;9(11):e113954.
- Kumar K, Sharma S, Kumar P, Deshmukh R. Therapeutic potential of GABA(B) receptor ligands in drug addiction, anxiety, depression and other CNS disorders. Pharmacol Biochem Behav. 2013;110:174–184.
- Paul AM, Branton WG, Walsh JG, et al. GABA transport and neuroinflammation are coupled in multiple sclerosis: regulation of the GABA transporter-2 by ganaxolone. Neuroscience. 2014;273:24–38.
- D’Amico E, Ziemssen T, Cottone S. To stop or not to stop disease modifying therapies in secondary progressive multiple sclerosis, that is the question. Expert Rev Neurother. 2017;17(9):847–849.
- Vesterinen HM, Connick P, Irvine CM, et al. Drug repurposing: a systematic approach to evaluate candidate oral neuroprotective interventions for secondary progressive multiple sclerosis. PLoS One. 2015;10(4):e0117705.
- Vicente Iturbe C, Ara Callizo JR, Huarte Lacunza R, et al. [Discontinuation and long-term adherence to beta interferon therapy in patients with multiple sclerosis]. Farm Hosp. 2012;36(2):77–83.
- Milo R, Miller A. Revised diagnostic criteria of multiple sclerosis. Autoimmun Rev. 2014;13(4–5):518–524.
- Wekerle H. Nature plus nurture: the triggering of multiple sclerosis. Swiss Med Wkly. 2015;145:w14189.
- Castel-Lacanal E, Tarri M, Loubinoux I, et al. Transcranial magnetic stimulation in brain injury. Ann Fr Anesth Reanim. 2014;33(2):83–87.
- Cui L. [Magnetic stimulation motor evoked potential in multiple sclerosis. Comparison with visual evoked potentials, brain stem auditory evoked potentials and somatosensory evoked potentials]. Zhonghua Shen Jing Jing Shen Ke Za Zhi. 1992;25(3):130–132, 189.
- Sahota P, Prabhakar S, Lal V, et al. Transcranial magnetic stimulation: role in the evaluation of disability in multiple sclerosis. Neurol India. 2005;53(2):197–201.
- Okamoto E, Ishikawa E, Yamamoto T, et al. Variability in amplitude and stimulation threshold values in motor evoked potential (MEP) monitoring during the resection of brain lesions. Clin Neurophysiol. 2015;126(6):1271–1278.
- Landazuri P, Eccher M. Simultaneous direct cortical motor evoked potential monitoring and subcortical mapping for motor pathway preservation during brain tumor surgery: is it useful? J Clin Neurophysiol. 2013;30(6):623–625.
- Mirdamadi JL, Suzuki LY, Meehan SK. Motor cortical plasticity in extrinsic hand muscles is determined by the resting thresholds of overlapping representations. Neuroscience. 2016;333:132–139.
- Chen R, Cros D, Curra A, et al. The clinical diagnostic utility of transcranial magnetic stimulation: report of an IFCN committee. Clin Neurophysiol. 2008;119(3):504–532.
- Temesi J, Gruet M, Rupp T, et al. Resting and active motor thresholds versus stimulus-response curves to determine transcranial magnetic stimulation intensity in quadriceps femoris. J Neuroeng Rehabil. 2014;11:40.
- Carson RG, Ruddy KL, McNickle E. What do TMS-evoked motor potentials tell us about motor learning? Adv Exp Med Biol. 2016;957:143–157.
- Pellicciari MC, Brignani D, Miniussi C. Excitability modulation of the motor system induced by transcranial direct current stimulation: a multimodal approach. Neuroimage. 2013;83:569–580.
- Horvath JC, Forte JD, Carter O. Evidence that transcranial direct current stimulation (tDCS) generates little-to-no reliable neurophysiologic effect beyond MEP amplitude modulation in healthy human subjects: a systematic review. Neuropsychologia. 2015;66:213–236.
- Udupa K, Chen R. Central motor conduction time. Handb Clin Neurol. 2013;116:375–386.
- Matsumoto H, Hanajima R, Terao Y, Ugawa Y. Magnetic-motor-root stimulation: review. Clin Neurophysiol. 2013;124(6):1055–1067.
- Lumsden DE, McClelland V, Ashmore J, et al. Central motor conduction time and diffusion tensor imaging metrics in children with complex motor disorders. Clin Neurophysiol. 2015;126(1):140–146.
- Farzan F, Barr MS, Hoppenbrouwers SS, et al. The EEG correlates of the TMS-induced EMG silent period in humans. Neuroimage. 2013;83:120–134.
- Chung SW, Rogasch NC, Hoy KE, Fitzgerald PB. Measuring brain stimulation inducedchanges in cortical properties using TMS-EEG. Brain Stimul. 2015;8(6):1010–1020.
- Hunter SK, McNeil CJ, Butler JE, et al. Short-interval cortical inhibition and intracortical facilitation during submaximal voluntary contractions changes with fatigue. Exp Brain Res. 2016;234(9):2541–2551.
- Hugos CL, Cameron MH. Assessment and measurement of spasticity in MS: state of the evidence. Curr Neurol Neurosci Rep. 2019;19(10):79.
- Centonze D, Koch G, Versace V, et al. Repetitive transcranial magnetic stimulation of the motor cortex ameliorates spasticity in multiple sclerosis. Neurology. 2007;68(13):1045–1050.
- Mori F, Codecà C, Kusayanagi H, et al. Effects of intermittent theta burst stimulation on spasticity in patients with multiple sclerosis. Eur J Neurol. 2010;17(2):295–300.
- Mori F, Ljoka C, Magni E, et al. Transcranial magnetic stimulation primes the effects of exercise therapy in multiple sclerosis. J Neurol. 2011;258(7):1281–1287.
- Boutière C, Rey C, Zaaraoui W, et al. Improvement of spasticity following intermittent theta burst stimulation in multiple sclerosis is associated with modulation of resting-state functional connectivity of the primary motor cortices. Mult Scler. 2017;23(6):855–863.
- Şan AU, Yılmaz B, Kesikburun S. The effect of repetitive transcranial magnetic stimulation on spasticity in patients with multiple sclerosis. J Clin Neurol. 2019;15(4):461–467.
- Iodice R, Dubbioso R, Ruggiero L, et al. Anodal transcranial direct current stimulation of motor cortex does not ameliorate spasticity in multiple sclerosis. Restor Neurol Neurosci. 2015;33(4):487–492.
- Korzhova J, Bakulin I, Sinitsyn D, et al. High-frequency repetitive transcranial magnetic stimulation and intermittent theta-burst stimulation for spasticity management in secondary progressive multiple sclerosis. Eur J Neurol. 2019;26(4):680–e44.
- Huang X, Stodieck SK, Goetze B, et al. Progressive maturation of silent synapses governs the duration of a critical period. Proc Natl Acad Sci U S A. 2015;112(24):E3131–E3140.